Діючі речовини: сунітиніб
SUTENT 12,5 мг твердих капсул
SUTENT 25 мг тверді капсули
SUTENT 37,5 мг твердих капсул
SUTENT тверді капсули 50 мг
Показання Чому використовується Сутент? Для чого це?
Сутент містить активну речовину сунітиніб, який є інгібітором протеїнкінази. Він використовується для лікування раку шляхом запобігання активності певної групи білків, які, як відомо, беруть участь у зростанні та поширенні ракових клітин.
Сутент буде призначений вам тільки лікарем, який має досвід застосування протипухлинних препаратів.
Сутент використовується для лікування дорослих з такими видами раку:
- Стромальний рак шлунково -кишкового тракту (GIST), тип раку шлунка та кишечника, у випадках, коли іматиніб (інший протипухлинний препарат) більше не діє або його більше не можна приймати.
- Метастатичний рак нирки (MRCC), тип раку нирки, який поширився на інші частини тіла.
- Нейроендокринні пухлини підшлункової залози (pNET) (пухлини клітин підшлункової залози, що продукують гормони), які прогресують або не піддаються резекції
. Якщо ви не впевнені, як діє Sutent або чому вам призначили цей препарат, зверніться до лікаря.
Протипоказання, коли Sutent не слід використовувати
Не приймайте Sutent:
- Якщо у вас алергія на сунітиніб або будь -який інший інгредієнт цього препарату (перерахований у розділі 6).
Заходи безпеки при застосуванні Що потрібно знати, перш ніж приймати Сутент
Скажіть своєму лікарю, перш ніж приймати Sutent:
- Якщо у вас високий кров'яний тиск. Сутент може спричинити підвищення артеріального тиску. Ваш лікар може перевірити артеріальний тиск під час прийому Сутенту, і за необхідності буде приймати ліки для зниження артеріального тиску.
- Якщо у вас є або були порушення крові, проблеми з кровотечею або синці. Лікування Sutent може нести підвищений ризик кровотечі, зміни кількості певних клітин крові, дефіцит яких призводить до анемії або впливає на здатність крові згортатися. Ризик кровотечі може бути більшим, якщо ви приймаєте варфарин або аценокумарол - ліки, що розріджують кров для запобігання утворенню тромбів. Скажіть своєму лікарю, якщо під час прийому Сутенту виникає кровотеча.
- Якщо у вас проблеми з серцем. Сутент може викликати проблеми з серцем. Повідомте свого лікаря, якщо ви відчуваєте сильну втому, у вас задишка або набрякають стопи та щиколотки.
- Якщо ви відчуваєте аномальні зміни серцевого ритму. Сутент може викликати зміни серцевого ритму. Під час лікування Сутентом лікар може зробити електрокардіограму, щоб оцінити ступінь цих змін. Скажіть своєму лікарю, якщо ви відчуваєте запаморочення, непритомність або ненормальне серцебиття під час прийому Сутенту.
- Якщо нещодавно у вас були проблеми із згустками крові у венах та / або артеріях (типи кровоносних судин), включаючи інсульт, інфаркт, емболію або тромбоз. Негайно зверніться до лікаря, якщо у вас виникнуть такі симптоми, як стискання або біль у грудях, біль у руках, спині, шиї або щелепі, задишка, оніміння або слабкість на одній стороні тіла, тремтіння при ходьбі, біль під час лікування Сутентом. Головний біль або запаморочення.
- Якщо у вас проблеми зі щитовидною залозою. Сутент може викликати проблеми з щитовидною залозою. Скажіть своєму лікарю, якщо ви легше втомлюєтесь під час прийому Сутенту, зазвичай відчуваєте холодніше, ніж інші люди, або ваш голос знижується. Перед прийомом Сутенту і регулярно під час прийому ліків слід перевіряти функцію щитовидної залози. Якщо щитовидна залоза не виробляє достатньо гормонів щитовидної залози, може знадобитися прийняти замісний гормон щитовидної залози.
- Якщо у вас є або були проблеми з підшлунковою залозою або жовчним міхуром. Повідомте свого лікаря, якщо у вас виникли будь -які з наведених нижче ознак та симптомів: біль у животі (у верхній частині живота), нудота, блювання та підвищення температури.Вони можуть бути викликані «запаленням підшлункової залози або жовчного міхура.
- Якщо у вас були або колись були проблеми з печінкою. Скажіть своєму лікарю, якщо під час лікування Сутентом ви відчуваєте будь -які з наведених нижче ознак та симптомів проблем із печінкою: свербіж, пожовтіння шкіри або очей, потемніння сечі та біль або дискомфорт у правій верхній частині живота. Ваш лікар повинен виконувати тести. для перевірки функції печінки до та під час лікування препаратом Сутент, а також відповідно до клінічних обставин.
- Якщо у вас є або були проблеми з нирками. Лікар контролюватиме роботу нирок.
- Якщо ви збираєтеся робити операцію або нещодавно перенесли операцію. Sutent може вплинути на спосіб загоєння ваших ран. Як правило, якщо ви збираєтесь зробити операцію, вам доведеться припинити використання Sutent. Ваш лікар вирішить, коли знову розпочати сутентне лікування.
- Перед початком лікування Сутентом бажано пройти огляд у стоматолога.
- якщо у вас болить рот, зуби та / або щелепа, набряк або виразки у роті, оніміння або відчуття тяжкості в щелепі або розхитування зубів, негайно повідомте про це лікаря та стоматолога.
- якщо ви проходите інвазивне лікування зубів або хірургію зубів, повідомте свого лікаря, що ви проходите лікування сутентом, особливо якщо ви також приймаєте внутрішньовенні бісфосфонати або приймали їх раніше. Бісфосфонати - це ліки, що використовуються для запобігання ускладненням з боку кісток, які могли бути призначені для іншої медичної проблеми.
- Якщо у вас є або колись були захворювання шкіри та підшкірної клітковини. "Гангренозна піодермія" (хвороблива виразка шкіри) або "некротизуючий фасцит" (швидко поширювана "інфекція шкіри / м'яких тканин, яка може бути смертельною) під час лікування цим препаратом. Припинення лікування. Серйозні шкірні реакції (Стівенс-Джонсон" Синдром, токсичний епідермальний некроліз, мультиформна еритема) були зареєстровані при застосуванні сунітинібу, який спочатку з’являвся на тулубі у вигляді червонуватих мішенеподібних плям або кругових плям, часто з пухирями в центрі. Реакція може прогресувати до поширеного утворення пухирів або лущення шкіри, а також може привести до летального результату. Якщо у вас з’явився висип або будь -який із цих шкірних симптомів, негайно зверніться до лікаря.
- Якщо у вас були або були судоми. Якнайшвидше повідомте лікаря, якщо у вас високий кров'яний тиск, головний біль, втрата зору.
- Якщо у вас діабет. У хворих на цукровий діабет слід регулярно перевіряти рівень цукру в крові, щоб побачити, чи потрібно змінювати дозу препаратів для діабету, щоб мінімізувати ризик зниження рівня цукру в крові.
Діти та підлітки
Сутент не показаний пацієнтам віком до 18 років. Сутент не вивчався у дітей та підлітків.
Взаємодії Які препарати або продукти харчування можуть змінити дію препарату Сутент
Повідомте свого лікаря або фармацевта, якщо ви приймаєте, нещодавно приймали або могли б приймати будь -які інші ліки, включаючи ті, що були придбані без рецепта та ті, що відпускаються без рецепта.
Деякі ліки можуть змінювати рівень сутенту в організмі. Вам слід повідомити лікаря, якщо ви приймаєте ліки, що містять такі активні речовини:
- кетоконазол, ітраконазол - використовується для лікування грибкових інфекцій
- еритроміцин, кларитроміцин, рифампіцин - застосовуються для лікування інфекцій
- ритонавір - використовується для лікування СНІДу
- дексаметазон - кортикостероїд, який використовується при кількох станах
- фенітоїн, карбамазепін, фенобарбітал - застосовуються для лікування епілепсії та інших неврологічних станів
- трав’яні препарати, що містять звіробій (Hypericum perforatum) - використовуються для лікування депресії та тривоги
Достатньо з їжею та напоями
Під час лікування Сутентом слід уникати вживання грейпфрутового соку.
Попередження Важливо знати, що:
Вагітність та годування груддю
Якщо ви вагітні або підозрюєте, що вагітні, повідомте про це свого лікаря.
Сутент не слід застосовувати під час вагітності, якщо це не є крайньою необхідністю. Ваш лікар обговорить з вами можливі ризики належного лікування під час вагітності.
Якщо вагітність можлива, ви повинні використовувати надійний метод контрацепції під час лікування Сутентом.
Якщо ви годуєте грудьми, повідомте про це свого лікаря. Не слід годувати грудьми під час лікування Сутентом.
Водіння автомобіля та роботу з машинами
Якщо ви відчуваєте запаморочення або незвичайну втому, будьте особливо обережні під час водіння автомобіля або роботи з механізмами.
Доза, спосіб та час введення Як застосовувати Сутент: Дозування
Завжди приймайте цей препарат точно так, як вам сказав ваш лікар.
Якщо є сумніви, зверніться до лікаря. Ваш лікар призначить правильну для вас дозу, виходячи з типу раку, який вам потрібно лікувати. Якщо ви лікуєтесь за ГІСТ або МРКК, звичайна доза становить 50 мг 1 раз на день, яку слід приймати протягом 28 днів (4 тижні), а потім 14 днів (2 тижні) відпочинку (без ліків), циклами по 6 тижнів. Якщо ви лікуєтесь від pNET, звичайна доза становить 37,5 мг один раз на день без періоду відпочинку. Ваш лікар визначить необхідну дозу і коли припинити належне лікування. Сутент можна приймати з їжею або без неї.
Передозування Що робити, якщо ви прийняли занадто багато Сутенту
Якщо Ви прийняли більше Сутенту, ніж слід
Якщо ви випадково прийняли занадто багато капсул, негайно зверніться до лікаря. Може знадобитися медична допомога
Якщо ви забули прийняти Сутент
Не приймайте подвійну дозу, щоб компенсувати пропущену дозу.
Побічні ефекти Які побічні ефекти Sutent
Як і всі ліки, цей препарат може викликати побічні ефекти, хоча вони виникають не у всіх.
Негайно зверніться до лікаря, якщо у вас виникли будь -які з цих серйозних побічних ефектів (див. Також Що потрібно знати, перш ніж приймати Сутент):
Проблеми з серцем. Повідомте свого лікаря, якщо ви відчуваєте сильну втому, у вас задишка або набрякають стопи та щиколотки. Це можуть бути симптоми серцевих проблем, таких як серцева недостатність та проблеми з серцевим м’язом (кардіоміопатія).
Проблеми з легенями або диханням. Скажіть своєму лікарю, якщо ви відчуваєте кашель, біль у грудях, раптову задишку або кашель крові. Це можуть бути симптоми легеневої емболії, яка виникає, коли тромби потрапляють до легенів.
Проблеми з нирками. Скажіть своєму лікарю, якщо ви відчуваєте "зміну частоти або відсутність сечовипускання, що може бути симптомом" ниркової недостатності.
Кровотеча. Негайно повідомте свого лікаря, якщо під час прийому Сутенту ви відчуваєте будь -який з наведених нижче симптомів або серйозну проблему з кровотечею: набряк, біль у животі (живіт); блювота з кров'ю; темний, липкий стілець; головний біль або зміни психічного стану, відкашлювання крові або мокротиння з кров’ю з легенів або дихальних шляхів.
Знищення пухлини, що спричиняє перфорацію кишечника. Скажіть своєму лікарю, якщо у вас сильний біль у кишечнику, температура, нудота, блювота, кров у стільці або змінили звички кишечника.
Інші побічні ефекти, які можуть виникнути при застосуванні Сутенту, це:
Дуже поширені побічні ефекти (можуть виникнути більш ніж у 1 з 10 осіб)
- Зменшення кількості тромбоцитів, еритроцитів та / або лейкоцитів (наприклад, нейтрофілів).
- Задишка
- Гіпертонія.
- Надмірна втома, занепад сил.
- Набряк, викликаний рідиною під шкірою та навколо очей, глибока алергічна висипка.
- Біль у роті / подразнення, хворобливість / запалення / сухість у роті, порушення смаку, розлад шлунка, нудота, блювота, діарея, запор, біль / набряк у животі, втрата / зниження апетиту.
- Зниження активності щитовидної залози (гіпотиреоз).
- Запаморочення
- Головний біль.
- Кровотеча з носа.
- Біль у спині, болі в суглобах.
- Болі в руках і ногах.
- Пожовтіння шкіри / зміна кольору шкіри, надмірна пігментація шкіри, зміна кольору волосся, висип на долонях і підошвах ніг, висип, сухість шкіри.
- Кашель.
- Лихоманка.
- Труднощі з засипанням.
Поширені побічні ефекти (можуть виникнути у 1-10 з 100 осіб)
- Утворення тромбів у кровоносних судинах.
- Недостатнє кровопостачання серцевого м’яза через обструкцію або звуження коронарних артерій.
- Біль у грудях.
- Зменшення кількості крові, що перекачується серцем.
- Затримка рідини також навколо легенів.
- Інфекції.
- Знижений рівень цукру в крові. Якщо ви відчуваєте ознаки та симптоми низького рівня цукру в крові: негайно повідомте лікаря, якщо відчуваєте втому, серцебиття, пітливість, голод та втрату свідомості.
- Втрата білка в сечі, що іноді призводить до набряку.
- Синдром грипу.
- Ненормальні аналізи крові, включаючи рівень ферментів печінки та підшлункової залози.
- Високий рівень сечової кислоти в крові.
- Геморой, ректальний біль, кровоточивість ясен, утруднене ковтання або неможливість ковтання.
- Печіння або хворобливі відчуття на мові, запалення слизової оболонки травного тракту, надлишок газів у шлунку або кишечнику.
- Втрата ваги.
- М'язово -скелетний біль (біль у м’язах і кістках), м’язова слабкість, м’язова втома, м’язовий біль, м’язові спазми.
- Сухість носа, закладеність носа.
- Надмірне сльозотеча.
- Зміни чутливості шкіри, сухість шкіри, свербіж, лущення та запалення шкіри, утворення пухирів, акне, зміна кольору нігтів, випадання волосся.
- Аномальні відчуття в кінцівках.
- Надмірне зменшення / збільшення чутливості, особливо на дотик.
- Печіння в животі.
- Зневоднення.
- Почервоніння обличчя.
- Зміна кольору сечі.
- Депресія.
- Озноб.
Нечасті побічні ефекти (можуть виникнути у 1-10 з 1000 осіб)
- Інфекції м’яких тканин, у тому числі в аногенітальній області, потенційно небезпечні для життя. Негайно зверніться до лікаря, якщо у вас виникли симптоми інфекції навколо рани шкіри, включаючи лихоманку, біль, почервоніння, набряк або відтік гною або крові.
- Інсульт.
- Інфаркт, викликаний перерваним або зниженим кровопостачанням серця.
- Зміни електричної активності серця або зміна серцевого ритму.
- Рідина навколо серця (перикардіальний випіт).
- Печінкова недостатність.
- Болі в животі (животі), викликані «запаленням підшлункової залози.
- Руйнування пухлини, що викликає перфорацію кишечника.
- Запалення (набряк і почервоніння) жовчного міхура з або без супутніх каменів.
- Ненормальний канал зв'язку між двома порожнинами тіла або зі шкірою.
- Біль у роті, зубах та / або щелепі, набряк або подразнення у роті, оніміння або відчуття тяжкості в щелепі або розхитування зубів. Це можуть бути ознаки та симптоми травми щелепи / щелепної кістки (остеонекроз). Негайно повідомте свого лікаря та стоматолога, якщо у вас виникнуть будь -які з цих ознак та симптомів.
- Надмірне вироблення гормонів щитовидної залози, що призводить до посилення метаболізму. Проблеми з загоєнням ран після операції.
- Збільшення м’язового ферменту в крові (креатинфосфокінази).
- Неправильна і надмірна реакція на алергени.
Рідкісні побічні ефекти (можуть виникнути у 1-10 з 10 000 осіб)
- Важкі реакції шкіри та / або слизових оболонок (синдром Стівенса-Джонсона, токсичний епідермальний некроліз, мультиформна еритема).
- Синдром пухлинного лізису (TLS) - TLS охоплює комплекс метаболічних ускладнень, які можуть виникнути під час лікування раку. Вони викликані продуктами розпаду уражених ракових клітин і можуть включати: нудоту, задишку, нерегулярне серцебиття, судоми м’язів, судоми, помутніння сечі та втому, пов’язані з аномальними результатами лабораторних досліджень (високий рівень калію, сечової кислоти та фосфорної кислоти) і низький рівень кальцію в крові), що може призвести до зміни функції нирок та гострої ниркової недостатності.
- Аномальний збій м’язів, що може спричинити проблеми з нирками (рабдоміоліз).
- Порушення функції мозку, що може призвести до різних симптомів, таких як головний біль, сплутаність свідомості, судоми та втрата зору (синдром задньої оборотної лейкоенцефалопатії).
- Хворобливі виразки шкіри (гангренозна піодермія).
- Запалення печінки (гепатит).
- Запалення щитовидної залози.
Повідомлення про побічні ефекти
Якщо Ви отримали будь -які побічні ефекти, зверніться до лікаря, це включає будь -які можливі побічні ефекти, не зазначені у цій брошурі. Ви також можете повідомляти про побічні ефекти безпосередньо за допомогою національної системи звітності, наведеної у Додатку V. Повідомляючи про побічні ефекти, ви можете допомогти надати більше інформації про безпеку застосування цього лікарського засобу.
Термін придатності та утримання
- Зберігайте цей препарат подалі від очей та недоступного для дітей місця.
- Не використовуйте цей препарат після закінчення терміну придатності, зазначеного на картонній упаковці та на етикетці після "EXP". Термін придатності відноситься до останнього дня місяця.
- Цей лікарський засіб не вимагає особливих умов зберігання.
- Не використовуйте цей препарат, якщо ви помітили, що упаковка пошкоджена або має ознаки втручання.
Не викидайте будь -які ліки через стічні води або побутові відходи. Запитайте у фармацевта, як викидати ліки, якими ви більше не користуєтесь. Це допоможе захистити навколишнє середовище.
Що містить Сутент
Сутент 12,5 мг твердих капсул
Діюча речовина - сунитиніб. Кожна капсула містить сунітинібу малату, що еквівалентно 12,5 мг сунітинібу.
Інші інгредієнти:
- Вміст капсули: маніт (Е421), кроскармелоза натрію, повідон (К-25) та стеарат магнію.
- Оболонка капсули: желатин, червоний оксид заліза (E172) та діоксид титану (E171).
- Чорнило: шелак, пропіленгліколь, гідроксид натрію, повідон та діоксид титану (Е171).
Сутентні тверді капсули по 25 мг
Діюча речовина - сунитиніб. Кожна капсула містить сунітинібу малату, еквівалентного 25 мг сунітинібу.
Інші інгредієнти:
- Вміст капсули: маніт (Е421), кроскармелоза натрію, повідон (К-25) та стеарат магнію.
- Оболонка капсули: желатин, діоксид титану (E171), жовтий оксид заліза (E172), червоний оксид заліза (E172), чорний оксид заліза (E172).
- Чорнило: шелак, пропіленгліколь, гідроксид натрію, повідон та діоксид титану (E171)
Сутентні тверді капсули 37,5 мг
Діюча речовина - сунитиніб. Кожна капсула містить сунітинібу малату, еквівалентного 37,5 мг сунітинібу.
Інші інгредієнти:
- Вміст капсули: маніт (Е421), кроскармелоза натрію, повідон (К-25) та стеарат магнію.
- Оболонка капсули: желатин, діоксид титану (E171), жовтий оксид заліза (E172).
- Чорнило: шелак, пропіленгліколь, гідроксид калію, чорний оксид заліза (Е172).
Сутентні тверді капсули 50 мг
Діюча речовина - сунитиніб. Кожна капсула містить сунітинібу малату, еквівалентного 50 мг сунітинібу.
Інші інгредієнти:
- Вміст капсули: маніт (Е421), кроскармелоза натрію, повідон (К-25) та стеарат магнію.
- Оболонка капсули: желатин, діоксид титану (E171), жовтий оксид заліза (E172), червоний оксид заліза (E172) та чорний оксид заліза (E172).
- Чорнило: шелак, пропіленгліколь, гідроксид натрію, повідон та діоксид титану (Е171).
Опис того, як виглядає Сутент, та вміст упаковки
Сутент 12,5 мг випускається у вигляді твердих желатинових капсул з помаранчевою кришкою та корпусом із написом "Pfizer" білими чорнилами на ковпачку та "STN 12,5 mg" на тілі, що містить кольорові гранули. Жовто-оранжевий.
Сутент 25 мг випускається у вигляді твердих желатинових капсул з карамельною кришкою та помаранчевим корпусом, з "Pfizer" білими чорнилами на ковпачку та "STN 25 mg" на тілі, що містить кольорові гранули. Жовто-оранжевий.
Сутент 37,5 мг випускається у вигляді твердих желатинових капсул з жовтою кришкою та корпусом із написом "Pfizer" чорними чорнилами на ковпачку та "STN 37,5 mg" на тілі, що містить кольорові гранули. Жовто-оранжевий.
Сутент 50 мг випускається у вигляді твердих желатинових капсул з кришкою та корпусом карамельного кольору, з "Pfizer" білими чорнилами на ковпачку та "STN 50 mg" на тілі, що містять жовто-оранжеві гранули. Випускається у флаконах по 30 капсул та перфорованих одиничних дозах, що містять 28 капсул по 1 капсулі.
Не всі розміри упаковок можна продавати.
Джерело з інформацією про упаковку: AIFA (Італійське агентство з лікарських засобів). Вміст, опублікований у січні 2016 р. Наявна інформація може бути не актуальною.
Щоб мати доступ до найновішої версії, бажано зайти на веб-сайт AIFA (Італійське агентство з лікарських засобів). Відмова від відповідальності та корисна інформація.
01.0 НАЗВА ЛЕКАРСТВЕННОГО ПРОДУКТУ
СУТЕНТ 12,5 МГ ЦВІРКІ КАПСУЛИ
02.0 ЯКІСНИЙ І КІЛЬКІСНИЙ СКЛАД
Кожна капсула містить сунітинібу малату, що відповідає 12,5 мг сунітинібу.
Повний список допоміжних речовин див. У розділі 6.1.
03.0 ФАРМАЦЕВТИЧНА ФОРМА
Тверда капсула.
Желатинові капсули з помаранчевою кришкою та корпусом, позначені білою фарбою "Pfizer" на кришці, "STN 12,5 мг" на тілі та містять жовто-оранжеві гранули.
04.0 КЛІНІЧНА ІНФОРМАЦІЯ
04.1 Терапевтичні показання
Стромальна пухлина шлунково -кишкового тракту (ГІСТ)
SUTENT призначається для лікування нерезектабельного та / або метастатичного раку шлунково -кишкового тракту (GIST) у дорослих після неефективності лікування іматинібом через резистентність або непереносимість.
Метастатичний нирково -клітинний рак (MRCC)
SUTENT призначений для лікування прогресуючої / метастатичної нирково -клітинної карциноми (MRCC) у дорослих.
Нейроендокринні пухлини підшлункової залози (pNET)
SUTENT призначений для лікування добре диференційованих, нерезекційних або метастатичних нейроендокринних пухлин підшлункової залози (pNETs) при прогресуючому захворюванні у дорослих.
Досвід застосування Сутенту як препарату першої лінії обмежений (див. Розділ 5.1).
04.2 Дозування та спосіб введення
Терапію сунітинібом повинен розпочинати лікар, який має досвід застосування протипухлинних засобів.
Дозування
Для GIST та MRCC рекомендована доза SUTENT становить 50 мг, які слід приймати перорально один раз на день, протягом 4 тижнів поспіль, після чого слід 2 тижні відпочинку (графік 4/2), щоб провести повний курс протягом 6 тижнів.
Для pNET рекомендована доза SUTENT становить 37,5 мг, яку слід приймати перорально один раз на день, без запланованого періоду відпочинку.
Корекція дози
Безпека і переносимість
Для GIST та MRCC можна змінювати дозу з кроком по 12,5 мг на основі індивідуальної безпеки та переносимості пацієнта. Добова доза не повинна перевищувати 75 мг, а також не повинна бути знижена нижче 25 мг.
Для pNET можна змінювати дозування з кроком 12,5 мг на основі індивідуальної безпеки пацієнта та переносимості. Максимальна доза, введена у дослідженні 3 фази pNET, становила 50 мг на добу.
Можливо, буде потрібно припинити прийом деяких доз, виходячи з безпеки та переносимості окремого пацієнта.
Інгібітори / індуктори CYP3A4
Слід уникати одночасного застосування сунітинібу з потужними індукторами CYP3A4, такими як рифампіцин (див. Розділи 4.4 та 4.5). Якщо це неможливо, можливо, доведеться збільшити дозу сунітинібу з кроком 12,5 мг (до 87,5 мг / день для GIST та MRCC або 62,5 мг / день для pNET) на основі ретельного моніторингу переносимості.
Слід уникати одночасного застосування сунітинібу з потужними інгібіторами CYP3A4, такими як кетоконазол (див. Розділи 4.4 та 4.5). Якщо це неможливо, можливо, доведеться зменшити дозу сунітинібу до мінімальної дози 37,5 мг на день для GIST та MRCC або 25 мг на день для pNET на основі ретельного моніторингу переносимості.
Слід розглянути вибір альтернативного супутнього лікарського засобу, який не має або має мінімальний потенціал для індукування або інгібування CYP3A4.
Особливі популяції
Педіатричне населення
Безпека та ефективність застосування сунітинібу у пацієнтів віком до 18 років не встановлені.
Немає даних.
Немає вказівок на специфічне застосування сунітинібу у дітей від народження до 6 років для лікування нерезектабельного та / або метастатичного ГІСТ після неефективності лікування иматінібом через резистентність або непереносимість. Немає вказівок на специфічне застосування сунітинібу у педіатричній популяції при лікуванні MRCC та при лікуванні добре диференційованих, нерезекційних або метастатичних pNET при прогресуванні захворювання.
Застосування сунітинібу у педіатричній популяції не рекомендується.
Пацієнти літнього віку (≥ 65 років))
Приблизно третина пацієнтів, які брали участь у клінічних випробуваннях та отримували сунітиніб, були у віці 65 років і старше. Ніяких значних відмінностей у безпеці та ефективності між молодими та літніми суб’єктами не спостерігалося.
Порушення функції печінки
При введенні сунітинібу пацієнтам з легкою або помірною печінковою недостатністю (стадії А та В за Чайлдом-П’ю) не потрібно коригувати початкову дозу. Застосування сунітинібу пацієнтам з тяжкою печінковою недостатністю (стадія С за Чайлдом-П’ю) не вивчалося, тому його застосування пацієнтам з печінковою недостатністю не рекомендується (див. Розділ 5.2).
Порушення функції нирок
Під час призначення сунітинібу пацієнтам з нирковою недостатністю (середньої та важкої форми) або термінальною стадією ниркової недостатності (ESRD), які проходять гемодіаліз, не потрібно коригувати початкову дозу. Подальші корекції дози слід проводити, виходячи з безпеки та переносимості пацієнтом (див. Розділ 5.2).
Спосіб введення
SUTENT призначений для прийому всередину. Його можна приймати з їжею або без неї.
Якщо доза не була прийнята, додаткову дозу приймати не слід. Наступного дня пацієнт повинен прийняти звичайну призначену дозу.
04.3 Протипоказання
Підвищена чутливість до активної речовини або до будь -якої з допоміжних речовин, перерахованих у розділі 6.1.
04.4 Спеціальні попередження та відповідні запобіжні заходи щодо використання
Слід уникати одночасного застосування з сильними індукторами CYP3A4, оскільки це може знизити концентрацію сунітинібу у плазмі крові (див. Розділи 4.2 та 4.5).
Слід уникати одночасного застосування з потужними інгібіторами CYP3A4, оскільки це може збільшити концентрацію сунітинібу у плазмі крові (див. Розділи 4.2 та 4.5).
Розлади шкіри та тканин
Зміна кольору шкіри, яка може бути обумовлена кольором активної речовини (жовтий), є дуже поширеною побічною реакцією, що виникає приблизно у 30% пацієнтів.Пацієнтів слід попередити, що під час лікування сунитинібом вони можуть змінити колір волосся або Інші можливі дерматологічні ефекти можуть включати сухість, потовщення або розтріскування шкіри, пухирі або випадкові висипання на долонях або підошвах.
Вищевказані реакції не були кумулятивними, були, як правило, оборотними і, як правило, не призводили до припинення лікування. Повідомлялося про випадки гангренозної піодермії, загалом оборотної після відміни препарату. Повідомлялося про серйозні шкірні реакції, включаючи випадки мультиформної еритеми ( ЕМ) та випадки, пов’язані з синдромом Стівенса-Джонсона (СЖС) та токсичним епідермальним некролізом (НЕТ), деякі з них летальні. Сунітиніб слід припинити. Якщо діагноз SJS або NET підтверджується, лікування не слід відновлювати. У деяких випадках з підозрою на ЕМ після усунення реакції пацієнти мали повторне введення терапії сунітинібом у нижчих дозах; деякі з цих пацієнтів також отримували супутнє лікування кортикостероїдами або антисептиками таміни.
Крововиливи та кровотечі, викликані пухлинами
Постмаркетинговий досвід повідомляв про геморагічні події, деякі з летальним результатом, включаючи шлунково-кишкові, дихальні, сечові та мозкові кровотечі.
Епізоди кровотеч траплялися у 18% пацієнтів, які отримували сунітиніб, порівняно з 17% пацієнтів у групі плацебо у дослідженні фази GIST 3. Пацієнти з MRCC на сунітинібі, які ніколи раніше не отримували сунітиніб, повідомили про випадки кровотечі у 39% випадків порівняно з 11% пацієнтів, які отримували IFN-α. Сімнадцять (4,5%) пацієнтів, які отримували сунітиніб, порівняно з 5 (1,7%) пацієнтами, які отримували IFN-α, зазнали епізодів кровотечі 3-го ступеня або вище. Двадцять шість відсотків пацієнтів, які отримували сунітиніб для цитокінорефрактерної МРКК, повідомили про епізоди кровотечі. Події кровотечі, за винятком носового кровотечі, мали місце у 21,7% пацієнтів, які отримували сунітиніб, порівняно з 9,85% пацієнтів у групі плацебо у дослідженні pNET фази 3. Рутинна оцінка цієї події повинна включати повний аналіз крові та фізичний огляд.
Епістаксис був найпоширенішою побічною геморагічною реакцією, про яку повідомлялося приблизно у половини пацієнтів із солідними пухлинами, які повідомляли про кровотечі.
Повідомлялося про події пухлинної кровотечі, іноді пов'язані з некрозом пухлини; деякі з цих кровотеч були смертельними.
У клінічних дослідженнях "пухлинна кровотеча сталася приблизно у 2% пацієнтів із ГІСТ. Ці події можуть виникнути раптово, а у разі раку легенів-у вигляді" важкого та небезпечного для життя кровохаркання або у вигляді легеневої кровотечі.Випадки легеневої кровотечі, деякі з летальним результатом, спостерігалися у клінічних випробуваннях, а також повідомлялося про постмаркетинговий період у пацієнтів, які отримували сунитиніб з приводу MRCC, GIST та раку легенів. SUTENT не схвалений для використання у пацієнтів з раком легенів.
Пацієнтів, які одночасно приймають антикоагулянти (наприклад, варфарин, аценокумарол), можна періодично контролювати шляхом повного аналізу крові (тромбоцитів), факторів згортання крові (PT / INR) та фізикального огляду.
Шлунково -кишкові розлади
Діарея, нудота / блювота, біль у животі, диспепсія та стоматит / біль у роті були найбільш поширеними побічними реакціями з боку шлунково -кишкового тракту; Також повідомлялося про випадки езофагіту (див. Розділ 4.8).
Підтримуюча допомога при шлунково-кишкових побічних реакціях, що потребують лікування, може включати ліки з протиблювотними, протидіарейними або антацидними властивостями.
Серйозні, іноді смертельні шлунково-кишкові ускладнення, включаючи перфорацію шлунково-кишкового тракту, мали місце у пацієнтів із внутрішньочеревною злоякісною пухлиною, які отримували лікування сунитинібом. У дослідженні GIST фази 3 смертельна шлунково -кишкова кровотеча сталася у 0,98% пацієнтів, які отримували плацебо.
Гіпертонія
Гіпертонія була дуже поширеною побічною реакцією, про яку повідомлялося в клінічних дослідженнях. Дозу сунітинібу зменшили або тимчасово призупинили приблизно у 2,7% пацієнтів, у яких виникла гіпертензія. У жодного з цих пацієнтів сунітиніб не припинився назавжди. систолічний або діастолічний 110 мм рт.ст.) спостерігався у 4,7% пацієнтів із солідними пухлинами.У МРКК та пацієнтів, які раніше не лікувалися, випадки артеріальної гіпертензії були зареєстровані у 33,9% пацієнтів на сунітинібі порівняно з 3,6% пацієнтів на ІФН-α. важка гіпертензія спостерігалася у 12% пацієнтів, які раніше не лікувалися, у групі сунітинібу та менш ніж у 1% пацієнтів у групі IFN-α. У дослідженні 3 фази pNET гіпертонія була зареєстрована у 26,5% пацієнтів, які приймали сунітиніб, порівняно з 4,9% пацієнтів у групі плацебо. Епізоди тяжкої гіпертензії траплялися у пацієнтів з pNET у 10%. Пацієнтів, які отримували сунітиніб та 3% пацієнти, які отримують плацебо. Тимчасове припинення лікування рекомендується пацієнтам з тяжкою неконтрольованою артеріальною гіпертензією, які приймають медикаментозне лікування. Лікування можна відновити, коли гіпертензію адекватно контролюють.
Гематологічні порушення
Абсолютне зменшення кількості нейтрофілів 3 та 4 ступенів спостерігалося у 10% та 1,7% пацієнтів, які брали участь у дослідженні GIST фази 3 відповідно, та у 16% та 1,6% пацієнтів, які були включені у дослідження. Фаза 3 MRCC та у 13 % та 2,4% пацієнтів, включених у дослідження фази 3 pNET. Зменшення кількості тромбоцитів 3 та 4 ступеня повідомлялося відповідно у 3,7% та 0,4% пацієнтів. Пацієнти, які брали участь у дослідженні GIST фази 3, у 8,2% та 1,1% пацієнтів, включених до дослідження фази 3 MRCC, та у 3,7% та 1,2% пацієнтів, які були включені до дослідження фази 3 щодо pNET.
Вищевказані події не були кумулятивними, були, як правило, оборотними і, як правило, не призводили до припинення лікування. включаючи крововиливи, пов'язані з тромбоцитопенією та нейтропенічними інфекціями.
Помічено, що анемія виникає як на ранніх, так і на пізніх стадіях лікування сунітинібом; повідомлялося про 3 та 4 ступінь.
На початку кожного циклу лікування пацієнтам, які отримують сунитиніб, слід провести повний аналіз крові.
Патології серця
У пацієнтів, які отримували сунитиніб, повідомлялося про серцево -судинні події, деякі з них з летальним результатом, включаючи серцеву недостатність, кардіоміопатію, ішемію міокарда та інфаркт міокарда. Ці дані вказують на те, що сунітиніб збільшує ризик кардіоміопатії. У пацієнтів, які проходили лікування, не було виявлено жодних додаткових факторів ризику кардіоміопатії, спричиненої сунітинібом, крім специфічної дії препарату.
У клінічних дослідженнях зменшення фракції викиду лівого шлуночка (LVEF) ≥ 20% і нижче нижньої межі норми відбувалося приблизно у 2% пацієнтів з ГІСТ, які отримували сунітиніб, у 4% пацієнтів з МРКК, що не піддається цитокінам, які отримували сунитиніб, і у 2 % пацієнтів з ГІСТ, які отримували плацебо. Ці зниження ФВЛН не видаються прогресивними і часто покращуються при продовженні лікування. У дослідженні, проведеному на пацієнтах з MRCC, які ніколи раніше не лікувалися, 27% пацієнтів, які отримували сунітиніб, і 15% пацієнтів, які отримували IFN-α, мали значення LVEF нижче нижньої межі норми. Двом пацієнтам (
У пацієнтів з ГІСТ епізоди «серцевої недостатності», «застійної серцевої недостатності» або «лівошлуночкової недостатності» були зареєстровані у 1,2% пацієнтів, які отримували сунитиніб та у 1% пацієнтів, які отримували плацебо. за даними GIST (n = 312), смертельні серцеві реакції, пов'язані з лікуванням, мали місце у 1% пацієнтів обох досліджуваних груп (сунітиніб та група плацебо). У 2-му етапі дослідження пацієнтів з цитокінорефрактерною МРКК 0,9 % пацієнтів повідомили про смертельний інфаркт міокарда, пов'язаний з лікуванням, а у фазі 3 дослідження пацієнтів з МРКК, які ніколи раніше не лікувалися,-0,6 % пацієнтів у групі IFN-α та 0% пацієнтів у групі сунітинібу повідомили про смертельні серцеві події. У дослідженні pNET фази 3 один пацієнт (1%), який приймав сунитиніб, мав смертельну серцеву недостатність, пов'язану з лікуванням. Можлива кореляція між пригніченням рецепторів тирозинкінази (РТК) та серцевою функцією неясна.
Пацієнти, які повідомили про серцеві події протягом 12 місяців до введення сунітинібу, такі як інфаркт міокарда (включаючи важку / нестабільну стенокардію), операцію на коронарному / периферичному обході, симптоматичну ХСН, цереброваскулярну подію або транзиторну ішемічну атаку або емболію легенів, були виключені з клінічні випробування із застосуванням сунітинібу. Невідомо, чи можуть пацієнти з такими супутніми станами мати підвищений ризик розвитку медикаментозної дисфункції лівого шлуночка.
Необхідно ретельно контролювати клінічні ознаки та симптоми застійної серцевої недостатності, особливо у пацієнтів з факторами серцевого ризику та / або в анамнезі з ішемічною хворобою серця.
Лікарі радять зважити цей ризик з потенційною користю препарату. Під час лікування сунитинібом за цими пацієнтами слід ретельно спостерігати за клінічними ознаками та симптомами застійної серцевої недостатності. Базові та періодичні оцінки фракції викиду лівого шлуночка також слід враховувати під час лікування пацієнта сунітинібом. У пацієнтів без серцевих факторів ризику все ще слід розглянути оцінку фракції викиду шлуночків на початковому рівні.
За наявності клінічних проявів ХСН рекомендується припинити лікування сунітинібом. Прийом сунітинібу слід перервати та / або зменшити дозу у пацієнтів без клінічних ознак застійної серцевої недостатності, але зі зменшенням фракції викиду між 20% та 50% від базового рівня.
Подовження інтервалу QT
Дані доклінічних досліджень (в пробірці І в природних умовах), проведені з дозами, вищими за рекомендовані для людей, свідчать про те, що сунітиніб може пригнічувати процеси реполяризації серця (наприклад, подовження інтервалу QT).
Збільшення інтервалу QTc до більш ніж 500 мсек відбувалося зі швидкістю 0,5%, а зміни від вихідного рівня більш ніж на 60 мсек відбувалися у 1,1% з 450 пацієнтів з солідними пухлинами; обидва ці параметри визнаються потенційно значущими варіаціями. При концентраціях, що відповідають приблизно дворазовим терапевтичним концентраціям, було виявлено, що сунітиніб подовжує інтервал QTcF (виправлений за формулою Фредеріки).
Подовження інтервалу QT вивчали у дослідженні, в якому брали участь 24 пацієнти у віці 20-87 років із прогресуючими злоякісними утвореннями.Результати цього дослідження продемонстрували, що сунітиніб впливає на інтервал QTc (визначається як скоригована середньо плацебо зміна> 10 мсек, з верхня межа 90% ДІ> 15 мсек) при терапевтичній концентрації (день 3) з використанням 24-годинного базового методу корекції, а при концентраціях вище до терапевтичних (день 9) з використанням обох методів корекції на початку. Жоден пацієнт не повідомив значення QTc> 500 мсек. Хоча вплив на інтервал QTcF спостерігався на 3-й день через 24 години після введення дози (тобто з очікуваною терапевтичною концентрацією у плазмі крові після рекомендованої початкової дози 50 мг) за допомогою 24-годинного базового методу корекції, клінічна значимість цього висновку незрозуміла .
Використовуючи комплексні серійні оцінки ЕКГ, що відповідають терапевтичному впливу або експозиції вище терапевтичного, у жодного з пацієнтів у оцінюваній або ІТТ популяції не спостерігалося подовження "важкого" інтервалу QTc (тому дорівнює або більше, ніж
3 клас CTCAE версії 3.0).
При терапевтичних концентраціях у плазмі середня максимальна зміна інтервалу QTcF (виправлена за формулою Фредеріки) від вихідного становила 9,6 мсек (90% ДІ 15,1 мсек). При терапевтичних концентраціях, що відповідають приблизно вдвічі терапевтичним концентраціям, максимальна зміна інтервалу QTcF від вихідного рівня становив 15,4 мс (90% ДІ 22,4 мс).
Моксифлоксацин (400 мг), використаний як позитивний контроль, демонстрував середню максимальну зміну інтервалу QTcF від вихідного рівня 5,6 мсек.
Подовження інтервалу QT може спричинити підвищений ризик шлуночкових аритмій, у тому числі Torsade de Pointes.
Сунітиніб слід з обережністю застосовувати пацієнтам з подовженням інтервалу QT, пацієнтам, які отримують антиаритмічні препарати, або лікарським засобам, які можуть подовжити інтервал QT, або пацієнтам з наявними відповідними захворюваннями серця, брадикардією або порушеннями. Одночасне застосування сунітинібу з потужними інгібіторами CYP3A4 має бути обмежене через можливе збільшення концентрації сунітинібу у плазмі (див. Розділи 4.2 та 4.5).
Венозні тромбоемболічні події
У клінічних дослідженнях, включаючи дослідження GIST та MRCC, приблизно у 1,0% пацієнтів із солідними пухлинами, які отримували сунітиніб, спостерігалися пов’язані з лікуванням венозні тромбоемболії.
У дослідженні GIST фази 3 у семи пацієнтів (3%), які отримували сунітиніб, і жодного пацієнта у групі плацебо не було венозних тромбоемболічних явищ; у п’яти із семи пацієнтів був тромбоз глибоких вен 3 ступеня (ТГВ), у двох - тромбоз глибоких вен 1 або 2 ступеня.
Тринадцять пацієнтів (3%), які отримували лікування сунітинібом у дослідженні 3 фази MRCC і ніколи раніше не отримували лікування, а чотири пацієнти (2%) з двох цитокінорефрактерних досліджень MRCC повідомили про венозні тромбоемболічні події. тромбоемболія легеневої артерії, у однієї з 2 ступеня та у восьмої з 4 ступеня. У восьми з цих пацієнтів була ТГВ, у однієї з 1 ступеня, у двох з 2 ступеня, у чотирьох з 3 та у 4 ступеня у пацієнта з легеневою емболією у дослідженні MRCC. , тугоплавкий до цитокінів, дозу припиняли.
З раніше нелікованих пацієнтів з МРКК, які отримували IFN-α, шість (2%) повідомили про венозні тромбоемболічні події, один пацієнт (емболія легеневої артерії, 4 ступінь).
У дослідженні фази 3 pNET повідомлялося про венозні тромбоемболічні події у 1 (1,2%) пацієнта у групі сунітинібу та 5 (6,1%) пацієнтів у групі плацебо. Двоє з цих пацієнтів, які отримували плацебо, повідомили про ТГВ, один 2-го та 3-го ступеня.
У ключових дослідженнях GIST, MRCC та pNET не було виявлено жодного випадку зі смертельним результатом. У постмаркетинговому періоді застосування препарату спостерігалися випадки зі смертельними наслідками (див. Дихальні події та розділ 4.8).
Артеріальні тромбоемболічні події
У пацієнтів, які отримували сунитиніб, повідомлялося про випадки артеріальних тромбоемболічних подій (АТЕ), іноді з летальним результатом. Найчастіші події включали порушення мозкового кровообігу, транзиторну ішемічну атаку та церебральну ішемію. До факторів ризику, пов'язаних з тромбоемболічними подіями в артеріях, на додаток до вже наявної злоякісної пухлини та віку 65 років і старше, належать гіпертонія, цукровий діабет та попередня тромбоемболічна подія.
Дихальні події
Пацієнти, у яких протягом останніх 12 місяців була емболія легеневої артерії, були виключені з клінічних досліджень із застосуванням сунітинібу.
У пацієнтів, які отримували сунітиніб у ключових дослідженнях фази 3, легеневі події (задишка, плевральний випіт, емболія легеневої артерії або набряк легенів) спостерігалися приблизно у 17,8% пацієнтів з ГІСТ, приблизно у 26,7% пацієнтів з МРКК та у 12% пацієнти з pNET.
Приблизно 22,2% пацієнтів із солідними пухлинами, включаючи GIST та MRCC, які отримували сунітиніб у клінічних дослідженнях, повідомили про легеневі події.
Випадки тромбоемболії легеневої артерії спостерігалися приблизно у 3,1% пацієнтів із ГІСТ та приблизно у 1,2% пацієнтів із МРКК, які отримували сунитиніб у дослідженнях фази 3 (див. Розділ 4.4. - Венозні тромбоемболічні події). у дослідженні фази 3. лікування сунітинібом у постмаркетингових умовах спостерігалося у рідкісних випадках зі смертельним результатом (див. розділ 4.8).
Порушення функції щитовидної залози
Рекомендується оцінка функції щитовидної залози шляхом вимірювання базових лабораторних показників у всіх пацієнтів. Пацієнтів з наявним гіпотиреозом або гіпертиреозом до початку лікування сунітинібом слід лікувати відповідно до стандартної клінічної практики. Під час лікування сунитинібом кожні 3 місяці слід проводити планову перевірку функції щитовидної залози. Крім того, під час лікування слід уважно стежити за всіма пацієнтами на предмет можливих ознак та симптомів дисфункції щитовидної залози, а пацієнтам, у яких з’являються будь -які ознаки та / або симптоми, що свідчать про порушення функції щитовидної залози, слід пройти лабораторне обстеження функції щитовидної залози відповідно до клінічних очікувань. Пацієнтів, у яких розвивається дисфункція щитовидної залози, слід лікувати відповідно до стандартної клінічної практики.
На початку або в кінці лікування сунітинібом спостерігався гіпотиреоз.
Повідомлялося про гіпотиреоз як побічну реакцію у 7 пацієнтів (4%), які отримували сунітиніб у двох дослідженнях MRCC, проведених у пацієнтів, що мають резистентність до цитокінів; у 61 пацієнта (16%), які отримували сунітиніб, і у трьох пацієнтів (
Крім того, повідомлялося про підвищення ТТГ у 4 пацієнтів (2%), які отримували лікування цитокінорефрактерної МРКК. В цілому 7% населення MRCC повідомили про клінічні або лабораторні ознаки гіпотиреозу, пов’язаного з лікуванням. Набутий гіпотиреоз спостерігався у 6,2% пацієнтів у дослідженні GIST, які приймали сунитиніб порівняно з 1% у групі плацебо. У дослідженні 3 фази pNET гіпотиреоз був зареєстрований у 6 пацієнтів (7,2%), які отримували сунітиніб, та у одного пацієнта (1,2%), який отримував плацебо.
Функцію щитовидної залози проспективно контролювали у двох дослідженнях у хворих на рак молочної залози; SUTENT не схвалений для лікування раку молочної залози. В одному дослідженні гіпотиреоз був зареєстрований у 15 пацієнтів (13,6%), які отримували сунітиніб, і у 3 суб’єктів (2,9%), які отримували стандартну терапію. Підвищення рівня ТТГ у крові було зареєстровано у 1 суб’єкта. (0,9%), який отримував сунітиніб, і ні у кого лікується стандартною терапією. Гіпертиреоз не повідомлявся у жодного з суб’єктів, які отримували сунітиніб, і був зареєстрований у 1 пацієнта (1,0%), який отримував стандартну терапію. суб'єкти (0,8%), які отримували капецитабін. Підвищення рівня ТТГ у крові було зареєстровано у 12 суб'єктів (5,0%), які отримували сунітиніб, і у жодного суб'єкта, який отримував капецитабін. Гіпертиреоз був зареєстрований у 4 пацієнтів (1,7%), які отримували сунітиніб, і у жодного суб’єкта, який отримував лікування капецитабіном. Зниження рівня ТТГ у крові було зареєстровано у 3 суб’єктів (1,3%), які отримували сунітиніб, і у жодного суб’єкта. 2 суб'єкти (0,8%) отримували сунітиніб та 1 суб'єкт (0,4%) отримували капецитабін. Підвищення Т3 було зареєстровано у 1 суб’єкта (0,8%), який отримував сунітиніб, і у жодного суб’єкта, який не отримував капецитабіну.
Випадки гіпертиреозу, деякі з яких слідували за гіпотиреозом, та випадки тиреоїдиту повідомлялися нечасто під час клінічних випробувань та на етапі збуту препарату.
Панкреатит
Підвищення активності ліпази та амілази в сироватці крові спостерігалося у пацієнтів з кількома солідними пухлинами, які отримували сунітиніб. Підвищення активності ліпази в сироватці крові було тимчасовим і, як правило, не асоціювалося з ознаками та симптомами панкреатиту у суб’єктів із солідними пухлинами різних типів.
Панкреатит спостерігався нечасто (
Повідомлялося про випадки серйозних випадків підшлункової залози, деякі з летальним результатом.
При появі симптомів панкреатиту лікування сунітинібом слід припинити, а пацієнтам надати відповідну підтримуючу допомогу. Під час дослідження фази 3 pNET не повідомлялося про випадки панкреатиту, пов’язані з лікуванням.
Гепатотоксичність
У пацієнтів, які отримували сунітиніб, спостерігалася гепатотоксичність. Випадки печінкової недостатності, іноді з летальним результатом, спостерігалися при функції печінки (аланін -трансаміназа [АЛТ], аспартат -трансаміназа [АСТ], рівень білірубіну). Ознаки або симптоми печінкової недостатності, лікування сунітинібом слід припинити, а пацієнтам - надати відповідну підтримуючу медичну допомогу.
Гепатобіліарні порушення
Лікування сунітінібом може бути пов'язане з холециститом, включаючи алітіазичний холецистит та енфматозний холецистит. У ключових клінічних дослідженнях частота холециститу становила 0,5%.
У постмаркетинговому періоді повідомлялося про випадки холециститу.
Ниркова функція
Повідомлялося про випадки ниркової недостатності, ниркової недостатності та / або гострої ниркової недостатності, у деяких випадках зі смертельним результатом.
Фактори ризику, пов'язані з нирковою недостатністю / недостатністю у пацієнтів, які отримували сунітиніб, включали, на додаток до вже існуючої нирково-клітинної карциноми: похилий вік, цукровий діабет, вже наявна ниркова недостатність, серцева недостатність, гіпертонія, сепсис, зневоднення / гіповолемія та рабдоміоліз.
Безпека продовження лікування сунітинібом у пацієнтів з протеїнурією помірного та тяжкого ступеня не оцінювалася систематично.
Повідомлялося про випадки протеїнурії та рідкісні випадки нефротичного синдрому. Рекомендується базовий аналіз сечі, і пацієнти повинні спостерігатися на предмет можливого розвитку або погіршення протеїнурії.
Пацієнтам з нефротичним синдромом лікування сунітинібом слід припинити.
Свищі
Якщо виникає свищ, лікування сунітинібом слід припинити. Існує обмежена інформація щодо тривалого лікування сунітинібом у пацієнтів із свищами.
Порушення процесу загоєння рани
Повідомлялося про випадки порушення загоєння ран під час терапії сунітинібом. Офіційних клінічних досліджень щодо впливу сунітинібу на загоєння ран не проводилося. З міркувань обережності рекомендується тимчасово припинити терапію сунітинібом у пацієнтів, які перенесли важку операцію. Клінічний досвід щодо термінів, необхідних для відновлення терапії після важкої операції, обмежений. Тому рішення про відновлення терапії сунітинібом після важкої операції має ґрунтуватися на клінічному судження про відновлення після операції.
Остеонекроз нижньої щелепи / верхньої щелепи
Повідомлялося про випадки остеонекрозу щелепи у пацієнтів, які отримували SUTENT. У більшості зареєстрованих випадків пацієнти проходили попереднє або супутнє лікування внутрішньовенними бісфосфонатами, при яких остеонекроз щелепи є ідентифікованим ризиком. Тому слід бути обережним при одночасному або комбінованому введенні СУТЕНТУ та внутрішньовенних бісфосфонатів.
Інвазивні стоматологічні втручання також були визначені як фактор ризику. Перед лікуванням препаратом СУТЕНТ слід продумати стоматологічний огляд та відповідну профілактичну стоматологічну допомогу. По можливості слід уникати інвазивних стоматологічних втручань у пацієнтів, які раніше приймали або приймають внутрішньовенні бісфосфонати (див. Розділ 4.8).
Гіперчутливість / ангіоневротичний набряк
Якщо виникає набряк унаслідок реакції гіперчутливості, лікування сунітинібом слід припинити та призначити стандартне медикаментозне лікування.
Розлади нервової системи
Порушення смаку
Про дисгевзію повідомлялося приблизно у 28% пацієнтів, які отримували сунітиніб під час клінічних випробувань.
Судоми
У клінічних випробуваннях із застосуванням сунітинібу та в постмаркетинговому періоді спостерігалися випадки судом у пацієнтів з рентгенологічними ознаками метастазів у мозок або без них. Крім того, повідомлялося про обмежену кількість повідомлень (головний біль, зниження пильності, порушення психічних функцій та втрату зору, у тому числі кортикальну сліпоту, слід контролювати медикаментозним лікуванням, включаючи контроль гіпертонії. Рекомендується тимчасова зупинка застосування сунітинібу; У цьому випадку лікування може бути відновлено на розсуд лікаря.
Синдром пухлинного лізису (TLS)
Випадки синдрому пухлинного лізису (ТЛС), деякі з летальним результатом, рідко спостерігалися у клінічних дослідженнях, і повідомлялося про постмаркетинговий період у пацієнтів, які отримували сунитиніб. Фактори ризику розвитку TLS включають високу пухлинну навантаження, наявну хронічну ниркову недостатність, олігурію, дегідратацію, гіпотензію та кислу сечу. За цими пацієнтами слід ретельно спостерігати та лікувати за клінічними показаннями; для цих пацієнтів слід розглянути можливість профілактичної гідратації.
Інфекції
Повідомлялося про випадки серйозних інфекцій з нейтропенією або без неї, включаючи деякі випадки зі смертельним результатом.
Інфекції, які найчастіше спостерігаються під час лікування сунітинібом, - це інфекції, характерні для пацієнта з раком, такі як дихальні шляхи, сечовивідні шляхи, шкіра та сепсис.
Повідомлялося про рідкісні, іноді з летальним результатом випадки некротизуючого фасцииту, включаючи промежину. Терапію сунітинібом слід припинити у пацієнтів, у яких розвивається некротичний фасціїт, і негайно розпочати відповідне лікування.
Гіпоглікемія
Були повідомлення про зниження рівня глюкози в крові під час лікування сунітинібом, деякі з них клінічно симптоматичні та потребують госпіталізації через втрату свідомості. У разі симптоматичної гіпоглікемії лікування сунітинібом слід тимчасово припинити. У хворих на цукровий діабет слід регулярно перевіряти рівень глюкози в крові, щоб оцінити, чи потрібно коригувати дозу препаратів для діабету, щоб мінімізувати ризик гіпоглікемії.
04.5 Взаємодія з іншими лікарськими засобами та інші форми взаємодії
Дослідження взаємодії проводилися тільки у дорослих.
Ліки, які можуть збільшувати плазмові концентрації сунітинібу
У здорових добровольців одночасне застосування одноразової дози сунітинібу з кетоконазолом, сильним інгібітором CYP3A4, призвело до збільшення комбінованих Cmax [сунітинібу + первинного метаболіту] та AUC0-∞ на 49% та 51% відповідно.
Введення сунітинібу з сильними інгібіторами CYP3A4 (наприклад, ритонавіром) , ітраконазол, еритроміцин, кларитроміцин, сік грейпфрута) може збільшити концентрацію сунитинібу.
Тому слід уникати комбінації з інгібіторами CYP3A4 або розглянути можливість застосування альтернативного лікарського засобу без або з мінімальним потенціалом інгібування CYP3A4.
Якщо це неможливо, можливо, доведеться зменшити дозу SUTENT мінімум до 37,5 мг / добу для GIST та MRCC або 25 мг / день для pNET на основі ретельного моніторингу переносимості (див. Розділ 4.2).
Ліки, які можуть зменшити плазмові концентрації сунітинібу
У здорових добровольців одночасне застосування одноразової дози сунітинібу та рифампіцину, індуктора CYP3A4, призвело до зменшення комбінованих Cmax [сунитинібу + первинного метаболіту] та AUC0-∞ на 23% та 46% відповідно.
Введення сунітинібу з сильними індукторами CYP3A4 (наприклад, дексаметазоном, фенітоїном, карбамазепіном, рифампіцином, фенобарбіталом або препаратами на основі трав, що містять звіробій /Hypericum perforatum) може знизити концентрацію сунітинібу. Тому слід уникати асоціації з індукторами CYP3A4 або розглядати альтернативний лікарський засіб, який не має або має мінімальний потенціал індукування CYP3A4. Якщо це неможливо, дозу SUTENT можна збільшити з кроком на 12, 5 мг до 87,5 мг / добу для GIST та MRCC або 62,5 мг / день для pNET) на основі ретельного моніторингу переносимості (див. розділ 4.2).
04.6 Вагітність та лактація
Вагітність
Не проводилося жодного дослідження у вагітних жінок, які отримували сунитиніб. Дослідження на тваринах показали репродуктивну токсичність, включаючи вади розвитку плода (див. Розділ 5.3).
SUTENT не слід застосовувати під час вагітності або жінкам, які не застосовують ефективні засоби контрацепції, якщо тільки потенційна користь не виправдовує потенційний ризик для плода. Якщо SUTENT використовується під час вагітності або якщо пацієнтка завагітніла під час лікування SUTENT, пацієнта слід поінформувати про потенційні ризики для плода.
Жінкам репродуктивного віку слід порадити використовувати ефективні засоби контрацепції та уникати вагітності під час лікування СУТЕНТОМ.
Час годування
Сунітініб та / або його метаболіти виділяються у молоко щурів. Невідомо, чи виділяється сунітиніб або його основний активний метаболіт у жіноче молоко. Оскільки активні речовини зазвичай виділяються з грудним молоком та враховуючи можливі серйозні побічні реакції у немовлят, які годують грудьми, жінкам не слід годувати грудьми під час лікування препаратом СУТЕНТ.
Родючість
Доклінічні дані свідчать про те, що фертильність чоловіків і жінок може погіршитися при лікуванні сунітинібом (див. Розділ 5.3).
04.7 Вплив на здатність керувати автомобілем та працювати з механізмами
Досліджень щодо впливу на здатність керувати транспортними засобами та працювати з механізмами не проводилося. Пацієнтів слід поінформувати про можливий розвиток запаморочення під час лікування сунитинібом.
04.8 Побічні ефекти
Короткий опис профілю безпеки
Найбільш серйозні побічні реакції, пов'язані з лікуванням сунітинібом, деякі з летальним результатом, це ниркова недостатність, серцева недостатність, емболія легеневої артерії, перфорація шлунково-кишкового тракту та крововиливи (наприклад, дихальні, шлунково-кишкові, пухлинні, кровотечі з сечі та мозку).
Найбільш поширені побічні реакції будь -якого ступеня (про які повідомляли пацієнти в ключових клінічних випробуваннях щодо РКС, GIST та pNET) включали зниження апетиту, порушення смаку, гіпертонію, втому, шлунково -кишкові розлади (наприклад, діарею, нудоту, стоматит, диспепсію та блювоту), зміну кольору синдрому шкіри та долонно-підошовної еритродизестезії. Ці симптоми можуть зменшитися при продовженні лікування. Під час лікування може виникнути гіпотиреоз. Гематологічні порушення (наприклад, нейтропенія, тромбоцитопенія та анемія) були одними з найпоширеніших побічних реакцій на лікарські засоби.
Смертельні несприятливі події, крім тих, що перераховані у розділі 4.4. або у розділі 4.8, які, як вважається, можливо пов’язані із сунітинібом, включають поліорганну недостатність, дисеміновану внутрішньосудинну коагуляцію, перитонеальну кровотечу, надниркову недостатність, пневмоторакс, шок та раптову смерть.
Таблиця побічних реакцій
Побічні реакції, про які повідомляли пацієнти з GIST, MRCC та pNET у об’єднаному наборі даних із 7115 пацієнтів, наведені нижче та класифіковані за класом системних органів, частотою та тяжкістю (NCI-CTCAE). Також повідомляється про постмаркетингові побічні реакції, виявлені у клінічних дослідженнях. У кожному класі частоти повідомляється про небажані ефекти у порядку їх зменшення.
Частоти визначаються таким чином: дуже часто (≥1 / 10), часто (≥1 / 100a
Таблиця 1 - Побічні реакції, про які повідомлялося у клінічних дослідженнях
Були згруповані такі терміни:
Назофарингіт та герпес ротової порожнини
b Бронхіт, інфекція нижніх дихальних шляхів, пневмонія та інфекція дихальних шляхів
c Абсцес, абсцес кінцівки, анальний абсцес, абсцес ясен, абсцес печінки, абсцес підшлункової залози, абсцес промежини, периректальний абсцес, абсцес прямої кишки, підшкірний абсцес та зубний абсцес
d Кандидоз стравоходу та кандидоз порожнини рота
і целюліт та шкірні інфекції
f Сепсис та септичний шок
g Абсцес живота, абдомінальний сепсис, дивертикуліт та остеомієліт
h Зниження апетиту та анорексія
i Дисгевзія, агевзія та зміна смаку
j Гострий коронарний синдром, стенокардія, нестабільна стенокардія, оклюзія коронарної артерії, ішемія міокарда
k Зменшення / аномалія фракції викиду
l Гострий інфаркт міокарда, інфаркт міокарда, тихий інфаркт міокарда
m Болі в ротоглотці та глотці
n Стоматит та афтозний стоматит
o Біль у животі, біль у нижній і верхній частині живота
p Перфорація шлунково -кишкового тракту та кишечника
q Холецистит та алітичний холецистит
r Пожовтіння шкіри, зміна кольору шкіри та порушення пігментації
s Псоріазиформний дерматит, ексфоліативна висип, висип, еритематозна висипка, фолікулярна висипка, генералізована висипка, макулярна висипка, макуло-папульозна висипка, папульозна висипка та свербіж
t Шкірна реакція та патологія шкіри
u Патологія нігтів та зміна кольору нігтів
v Втома та астенія
w Набряк обличчя, набряки та периферичні набряки
x амілаза та підвищена амілаза
* Включає смертельні події
Опис виявлених побічних реакцій
Інфекції та інвазії: Повідомлялося про випадки серйозних інфекцій (з нейтропенією або без неї), включаючи випадки зі смертельним результатом. Повідомлялося про випадки некротизуючого фасцииту, іноді з летальним результатом, який також може вплинути на область промежини (див. Також розділ 4.4).
Порушення з боку крові та лімфатичної системи: Повідомлялося про випадки тромботичної мікроангіопатії. У цих випадках рекомендується тимчасове призупинення SUTENT; після усунення цих подій лікування може бути відновлено на розсуд лікаря.
Порушення з боку імунної системи: Повідомлялося про реакції гіперчутливості, включаючи ангіоневротичний набряк.
Порушення з боку нервової системи: Повідомлялося про декілька випадків, деякі з летальним результатом, у пацієнтів із судомами та рентгенологічними ознаками реверсивної задньої синдрому лейкоенцефалопатії (RPLS) (див. Також розділ 4.4).
Порушення обміну речовин і харчування: Повідомлялося про більш високу частоту гіпоглікемічних подій у пацієнтів з pNET порівняно з пацієнтами з MRCC та GIST. Однак багато побічних явищ, які спостерігалися у клінічних випробуваннях, не вважалися пов'язаними з досліджуваним лікуванням.
Гепатобіліарні порушення: Повідомлялося про печінкову дисфункцію, яка може включати порушення функцій печінки, гепатит або печінкову недостатність.
З боку шкіри та підшкірної клітковини: Повідомлялося про випадки гангренозної піодермії, які зазвичай були оборотними після припинення лікування (див. Також розділ 4.4).
З боку опорно -рухового апарату та сполучної тканини: Повідомлялося про випадки міопатії та / або рабдоміолізу, деякі з яких пов’язані з гострою нирковою недостатністю. Пацієнтів з ознаками або симптомами м’язової токсичності слід лікувати відповідно до стандартної медичної практики.
Повідомлялося про випадки утворення нориць, іноді пов'язаних з некрозом пухлини та регресією, у деяких випадках з летальним результатом.
Повідомлялося про випадки остеонекрозу щелепи у пацієнтів, які отримували SUTENT, багато з яких траплялося у пацієнтів, у яких були визнані фактори ризику остеонекрозу щелепи, особливо вплив внутрішньовенних бісфосфонатів та / або захворювання стоматології в анамнезі, що потребує інвазивних стоматологічних втручань (див. також розділ 4.4).
Повідомлення про підозрювані побічні реакції
Повідомлення про підозрювані побічні реакції, що виникають після реєстрації лікарського засобу, є важливими, оскільки вони дозволяють здійснювати постійний моніторинг співвідношення користь / ризик лікарського засобу. Медичних працівників просять повідомляти про будь -які підозрювані побічні реакції через національну систему звітності у "Додатку V" .
04.9 Передозування
Специфічного антидоту для передозування сунітинібом немає, і лікування передозування повинно включати загальні підтримуючі заходи. Якщо є показання, усунення абсорбованої активної речовини може бути досягнуто блювотою або промиванням шлунка. Повідомлялося про випадки передозування; у деяких випадках у цих випадках побічні реакції, що мали місце, узгоджувалися з відомим профілем безпеки застосування сунітинібу.
05.0 ФАРМАКОЛОГІЧНІ ВЛАСТИВОСТІ
05.1 Фармакодинамічні властивості
Фармакотерапевтична група: протипухлинні засоби, інгібітори протеїнкінази.
Код ATC: LO1XE04.
Механізм дії
Сунітініб пригнічує рецептори множинної тирозинкінази (РТК), які беруть участь у зростанні пухлини, пухлинному неоангіогенезі та метастатичному прогресуванні раку. Сунітініб був ідентифікований як інгібітор рецепторів фактора росту тромбоцитів (PDGFRα та PDGFRβ), рецепторів судинного ендотеліального фактора росту (VEGFR1, VEGFR2 та VEGFR3), рецептора фактора стовбурових клітин (KIT), рецептора тирозинкінази FLT3 (Fms-подібний тирозинкіназа 3), рецептор CSF-1R (рецептор фактора, що стимулює колонії (CSF-1R)) та рецептор нейрофічного фактора, що походить від глії (RET). У біохімічних та клітинних тестах основний метаболіт демонструє ефективність, порівнянну з сунітинібом.
Клінічна ефективність та безпека
Безпеку та клінічну ефективність сунітинібу вивчали при лікуванні пацієнтів з ГІСТ, стійкими до іматинібу (тобто пацієнтами, які прогресували під час або після лікування іматинібом) або непереносимістю до іматинібу (тобто тих, хто мав значну токсичність під час лікування іматинібом) які запобігали продовженню лікування), при лікуванні пацієнтів з MRCC та при лікуванні пацієнтів з неоперабельною pNET.
Ефективність залежить від часу до прогресування пухлини та збільшення виживаності у пацієнтів із ГІСТ, виживання без прогресування та об’єктивної відповіді у пацієнтів, які раніше не лікувались МРКК та МРКК, що не піддаються терапії відповідно до цитокінів МРКК, та виживання у пацієнтів без прогресування. з pNET.
Стромальні пухлини шлунково -кишкового тракту (ГІСТ)
Початкове відкрите ескалаційне дослідження було проведено у пацієнтів з ГІСТ після невдачі іматинібу (середня максимальна добова доза 800 мг) через резистентність або непереносимість. Було зараховано 97 пацієнтів з різними дозуваннями та режимами дозування; 55 пацієнтів лікувалися препаратом Сутент 50 мг відповідно до рекомендованої 4-тижневої дози та двотижневої схеми відміни препарату (графік 4/2).
У цьому дослідженні середній час до прогресування TTP становив 34,0 тижня (95% ДІ = 22,0-46,0 тижнів).
Рандомізоване, подвійне сліпе, плацебо-контрольоване дослідження фази 3 сунітинібу проводилося у пацієнтів з ГІСТ, які мали непереносимість або мали прогресування захворювання під час або після лікування іматинібом (середня максимальна добова доза 800 мг). У цьому дослідженні 312 пацієнтів (2: 1) були рандомізовані для лікування 50 мг сунітинібу або плацебо перорально один раз на день відповідно до схеми 4/2 до прогресування захворювання або припинення дослідження з „іншої причини (207 пацієнтів отримали сунітініб та 105 плацебо).
Первинною кінцевою точкою ефективності дослідження була TTP, визначена як час від рандомізації до першого документування об’єктивної прогресії пухлини.На момент попередньо визначеного проміжного аналізу середня TTP із сунітинібом становила 28,9 тижня (95% ДІ = 21,3-34,1 тиждень ) за оцінкою дослідника та 27,3 тижня (95% ДІ = 16,0-32,1 тижня) за оцінкою Незалежної оглядової комісії і статистично перевищувала 5,1-тижневу ТТР, отриману з плацебо (95% ДІ = 4,4-10,1 тижня, стор
Незалежний оглядовий комітет. Різниця в загальній виживаності статистично була на користь сунітинібу [коефіцієнт небезпеки: 0,491 95% (ДІ 0,290-0,831)]; ризик смерті був у 2 рази вищим у пацієнтів, які перебували на групі плацебо, ніж на групі сунітинібу.
Після проміжного аналізу ефективності та безпеки, за рекомендацією незалежного DSMB, дослідження було сліпим, і пацієнтам, які перебували у групі плацебо, було запропоновано перейти на відкрите лікування сунітинібом.
Загалом 255 пацієнтів проходили лікування сунітинібом на відкритому етапі лікування, у тому числі 99 пацієнтів, які спочатку отримували плацебо.
Аналіз первинних та вторинних кінцевих точок на відкритій фазі дослідження підтвердив результати, отримані під час проміжного аналізу, як показано в таблиці нижче.
Таблиця 2 - Резюме кінцевих показників ефективності (популяція ITT)
Середня загальна виживаність (ОС) у популяції ІТТ становила 72,7 тижня та 64,9 тижня (ЧСС 0,876, 95% ДІ 0,679 - 1,129, р = 0,306) відповідно у групі лікування сунитинібом та плацебо відповідно. У цьому аналізі група плацебо включала пацієнтів, рандомізованих до плацебо, які згодом перейшли на відкрите лікування сунітинібом.
Метастатичний нирково -клітинний рак (MRCC) у пацієнтів, які раніше не лікувалися
Було проведено рандомізоване, багатоцентрове, міжнародне дослідження Фази 3 для оцінки ефективності та безпеки застосування сунітинібу порівняно з інтерфероном IFN-α у пацієнтів, які раніше не лікувалися МРКК. повторювані 6-тижневі цикли. Кожен цикл складається з 4 тижнів по 50 мг на добу перорально, потім 2 тижні без прийому препарату (схема 4/2) або з пероральним підшкірним введенням IFN-α у дозі 3 млн одиниць (МО ) перший тиждень, 6 МО на другий тиждень і починаючи з третього тижня в дозі 9 МО відповідно до курсу лікування 3 дні без послідовності кожного тижня.
Середня тривалість лікування становила 11,1 місяця (діапазон: 0,4 - 46,1) для лікування сунітинібом та 4,1 місяця (діапазон 0,1 - 45,6) для лікування IFN -α. Серйозні побічні реакції (TRSAE), пов'язані з лікуванням, були зареєстровані у 23,7% пацієнтів, які отримували сунітиніб, та у 6,9% пацієнтів, які отримували IFN-α. Однак коефіцієнт припинення застосування через побічні ефекти становив 20% для сунітинібу та 23% для IFN-α. Припинення лікування відбулося у 202 пацієнтів (54%) у групі сунітинібу та 141 пацієнта (39%) у групі IFN-α.
Зниження дози сталося у 194 пацієнтів (52%), які отримували сунітиніб, та 98 пацієнтів (27%), які отримували IFN-α. Пацієнтів лікували до прогресування захворювання або припинення дослідження. Первинною кінцевою точкою ефективності було виживання без прогресування захворювання (PFS).
Плановий проміжний аналіз показав статистично значущу перевагу для SUTENT над IFN-α. У цьому дослідженні середня PFS для групи сунітинібу становила 47,3 тижня порівняно з 22,0 тижня для групи IFN-α; коефіцієнт небезпеки становив 0,415 (95% ДІ: 0,320-0,539, р-значення
Лікування сунітинібом асоціювалося з тривалістю виживання, ніж лікування IFN-α. Середня загальна виживаність становила 114,6 тижнів для групи сунітинібу (95% ДІ: 100,1 - 142,9 тижнів) та 94,9 тижня для групи ІФН -α (95% ДІ: 77,7 - 117,0 тижнів) з коефіцієнт небезпеки 0,821 (95% ДІ: 0,673-1,001; р = 0,0510 на основі нестратифікованого логарифмічного тесту).
Виживаність без прогресування захворювання та загальна виживаність (ОС), які спостерігаються з метою лікування популяції (ІТТ) та визначаються за допомогою рентгенологічної лабораторної оцінки, узагальнені у наступних таблицях:
Резюме кінцевих показників ефективності (популяція ITT)
Цитокін-рефрактерна метастатична нирково-клітинна карцинома (MRCC)
Було проведено дослідження другої фази з SUTENT у пацієнтів, що не піддавалися попередній терапії цитокінами, які отримували інтерлейкін-2 або IFN-α. Шістдесят три пацієнти отримували пероральну початкову дозу сунітинібу 50 мг один раз на день протягом 4 тижнів поспіль, а потім 2-тижневий період відпочинку для завершення повного 6-тижневого курсу (схема лікування 4/2). Первинною кінцевою точкою ефективності була об'єктивна швидкість відповіді (Об’єктивно
Частота відповідей (ORR)) відповідно до критеріїв RECIST (Критерії оцінки відповідей у твердих пухлинах).
У цьому дослідженні об’єктивний рівень відповіді становив 36,5% (95% ДІ 24,7% -49,6%), а середній час до прогресування (ТТП) становив 37,7 тижнів (95% ДІ 24,0-46,4 тижня).
Відкрите, одноцентрове, багатоцентрове підтверджуюче дослідження для оцінки ефективності та безпеки застосування сунітинібу було проведено у пацієнтів з МРКК, що не піддається попередній цитокіновій терапії. Сто шість пацієнтів отримували принаймні одну дозу 50 сунитинібу. Мг рамки схеми 4/2.
Первинною кінцевою точкою ефективності цього дослідження була швидкість ОРР. Вторинні кінцеві точки включали TTP, тривалість відповіді (DR) та загальну виживаність (OS).
У цьому дослідженні ORR становив 35,8% (95% ДІ 26,8% -47,5%). DR та середня ОС ще не були досягнуті.
Нейроендокринні пухлини підшлункової залози (pNET)
Відкрите, багатоцентрове, фаза 2 підтримуючого дослідження оцінила ефективність та безпеку монотерапії сунітинібом у дозах 50 мг на день за схемою 4/2 [4 тижні лікування, 2 тижні відпустки] у пацієнтів з непрацездатним pNET У когорті 66 у пацієнтів з раком острівцевих клітин підшлункової залози кінцева точка первинної відповіді становила 17%.
Було проведено ключове фазове, багатоцентрове, міжнародне, рандомізоване, подвійне сліпе, плацебо-контрольоване дослідження монотерапії сунітинібом у пацієнтів з неоперабельною pNET.
Пацієнти, яким необхідно було задокументовано прогресування захворювання на основі RECIST протягом останніх 12 місяців, були рандомізовані (1: 1) для отримання 37,5 мг сунітинібу один раз на день без запланованого періоду відміни (n = 86) або плацебо (n = 85) .
Первинною кінцевою точкою була оцінка виживання без прогресування (ПФС) у пацієнтів, які приймали сунітиніб, порівняно з тими, хто отримував плацебо. Іншими кінцевими точками були ОС, відсоток ОРР, результати, повідомлені пацієнтами) та безпека.
З точки зору демографічних характеристик, групи пацієнтів, які отримували сунитиніб та плацебо, були порівнянними. Крім того, 49% пацієнтів, які отримували сунітиніб, і 52% пацієнтів, які отримували плацебо, мали нефункціонуючі пухлини, а 92% пацієнтів на обох руках мали метастази в печінку. Дослідження дозволило використовувати аналоги соматостатину. 66% пацієнтів, які отримували сунітиніб та 72% пацієнтів, які отримували плацебо, раніше отримували системну терапію. Крім того, 24% пацієнтів у групі сунітинібу та 22% пацієнтів у плацебо Група отримувала аналоги соматостатину. Клінічно значуща перевага сунітинібу ФФС порівняно з плацебо була відзначена під час оцінки дослідником. Середня ПФС становила 11,4 місяця у групі сунітинібу порівняно з 5,5 місяців у групі плацебо [коефіцієнт ризику: 0,418 (95% ДІ 0,263 , 0,662), p-значення = 0,0001]; подібні результати були помічені, коли оцінки реакції пухлини, засновані на застосуванні RECIST до вимірювань пухлини, проведених дослідниками, були використані для визначення прогресування захворювання, як показано в таблиці 3. A коефіцієнт небезпеки на користь сунітинібу спостерігався у всіх підгрупах, оцінених за вихідними характеристиками, включаючи аналіз за кількістю попередніх системних терапій. Всього 29 пацієнтів у групі сунітинібу та 24 у групі плацебо не отримували попередньої системної терапії; у цих пацієнтів там »коефіцієнт небезпеки для PFS це було 0,365 (95% ДІ 0,156, 0,857), р = 0,0156.
Подібним чином серед 57 пацієнтів у групі сунітинібу (у тому числі 28 з 1 попередньою системною терапією та 29 з 2 або більше системними терапіями) та 61 пацієнт у групі плацебо (у тому числі 25 з 1 попередньою системною терапією та 36 з 2 або більше системними терапіями) ) l "коефіцієнт небезпеки для PFS це було 0,456 (95% ДІ 0,264, 0,787), р = 0,0036.
Аналіз чутливості PFS проводили, коли PFS ґрунтувався на вимірах пухлини, проведених дослідниками, і коли всі суб’єкти, піддані цензурі з інших причин, ніж припинення дослідження, вважалися подіями PFS. Цей аналіз дав консервативну оцінку ефекту від лікування сунитинібом та підтвердив первинний аналіз, продемонструвавши "коефіцієнт небезпеки 0,507 (95% ДІ 0,350, 0,733), р = 0,000193. Основне дослідження підшлункової NET було припинено достроково на підставі рекомендації незалежного Комітету з оцінки лікарських засобів, а основна кінцева точка базувалася на оцінці дослідника: обидві умови могли вплинути на оцінку ефекту лікування.
Щоб виключити упередженість в оцінці дослідниками ПФС, був проведений незалежний, сліпий центральний огляд діагностичних зображень; цей огляд підтримав оцінку слідчого, як показано в таблиці 3.
Таблиця 3 - Результати ефективності дослідження фази 3 pNET
CI = довірчий інтервал, HR = коефіцієнт небезпеки, NA = Не застосовується, NR = не досягнуто
двосторонній нестратифікований тест логарифмічного рангу
b Точний тест Фішера
Загальна цифра виживання не була дозрілою під час проведення аналізу. Було 9 випадків смерті у групі сунітинібу та 21 смерть у групі плацебо. Статистично значуща різниця спостерігалася у об’єктивній відповіді на сунітиніб порівняно з плацебо.
У випадках прогресування захворювання пацієнтів повідомляли про лікування, яке вони отримували, а пацієнтів, які приймали плацебо, пропонували включити до іншого відкритого подовженого дослідження із застосуванням сунітинібу. Через раннє закриття дослідження решта пацієнтів були поінформовані про лікування, яке вони отримували, і їм було запропоновано доступ до відкритого подовженого дослідження із застосуванням сунітинібу. Загалом у дослідженні 59 пацієнтів у групі плацебо отримували сунітиніб. розширення.
Результати опитувальника якості життя Європейської організації з дослідження та лікування раку (EORTC QLQ-C30) показали, що загальна якість життя, пов'язана зі здоров'ям, і п'ять функціональних областей (фізична, рольова, когнітивна, емоційна та соціальні) були збережені у пацієнтів, які отримували сунітиніб, порівняно з тими, хто отримував плацебо, з незначною кількістю симптоматичних побічних ефектів.
Педіатричне населення
Європейське агентство з лікарських засобів відстрочило зобов'язання надсилати результати досліджень з SUTENT в одну або кілька підгруп педіатричної популяції для лікування ГІСТ (див. Розділ 4.2 для інформації про застосування у педіатрії).
Європейське агентство з лікарських засобів відмовилося від зобов’язання подавати результати досліджень з SUTENT у всіх підгрупах педіатричної популяції для лікування ниркових та ниркових карцином тазу (за винятком нефробластоми, нефробластоматозу, саркоми, клітин саркоми, мезобластичної нефроми, ниркової мозкової оболонки) карцинома та рабдоїдна пухлина нирки) (див. розділ 4.2 для отримання інформації про застосування у педіатрії).
Європейське агентство з лікарських засобів скасувало зобов’язання подавати результати досліджень з SUTENT у всіх підгрупах педіатричної популяції для лікування гастроентеропанкреатичних нейроендокринних пухлин (за винятком нейробластоми, нейрогангліобластоми, феохромоцитоми) (див. Розділ 4.2 для інформації про застосування у педіатрії ).
05.2 "Фармакокінетичні властивості
Фармакокінетику сунітинібу оцінювали у 135 здорових добровольців та у 266 пацієнтів із солідними пухлинами. Фармакокінетика була однаковою у всіх досліджених популяціях пацієнтів із солідними пухлинами та у здорових добровольців.
У режимах прийому 25-100 мг площа під кривою (AUC) та Cmax збільшуються пропорційно дозі. При повторному щоденному введенні сунітиніб накопичується у 3-4 рази, а основний активний метаболіт-у 7-10 разів. Рівноважна концентрація сунітинібу та його основного активного метаболіту досягається протягом 10-14 днів. До 14 -го дня загальна концентрація сунітинібу в плазмі крові та його основного активного метаболіту становить 62,9 - 101 нг / мл і являє собою прогнозовані цільові концентрації на основі доклінічних даних для інгібування фосфорилювання рецепторів. в пробірці і призводять до зменшення пухлинного застою / росту в природних умовах.
Основний активний метаболіт становить 23-37% від загальної експозиції лікарського засобу. При повторних курсах прийому один раз на день або повторних курсах дозування не спостерігається значних змін у фармакокінетиці сунітинібу або основного активного метаболіту.
Поглинання
Після перорального введення сунітинібу пікові концентрації (Cmax) зазвичай спостерігаються протягом 6-12 годин (Tmax) після прийому препарату.
Їжа не впливає на біодоступність сунітинібу.
Розповсюдження
Зв’язування сунітинібу та його основного активного метаболіту з білками плазми людини у тестах в vitro вони становили відповідно 95% та 90%, без «очевидної залежності від концентрації».
Очевидний об'єм розподілу (Vd) сунітинібу був великим - 2230 л, що вказує на розподіл у тканинах.
Метаболічні взаємодії
Розрахункові значення Ki в пробірці для всіх досліджених ізоформ цитохрому (CYP) (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 / 5 та CYP4A9 / 11) малоймовірно, що сунітініб та його основний клінічно активний метаболіт a метаболізм інших активних речовин, які можуть метаболізуватися цими ферментами.
Біотрансформація
Сунітиніб переважно метаболізується CYP3A4, ізоформою цитохрому Р450, яка продукує його основний активний метаболіт - десетилсунітиніб, який далі метаболізується цим самим ізоферментом.
Слід уникати одночасного застосування сунітинібу та сильних індукторів або інгібіторів CYP3A4, оскільки рівень сунітинібу в плазмі крові може бути змінений (див. Розділи 4.4 та 4.5).
Ліквідація
Екскреція відбувається переважно з калом (61%), а незмінена діюча речовина та її метаболіти нирками виводять 16% введеної дози. Сунітініб та його основний активний метаболіт були основними сполуками, виявленими у плазмі крові, сечі та фекаліях та становили відповідно 91,5%, 86,4%та 73,8%радіоактивності, виявленої у об’єднаних зразках. Незначні метаболіти були виявлені в сечі та фекаліях, але зазвичай не виявляються у плазмі. Загальний оральний кліренс (CL / F) становив 34-62 л / год.
Після перорального застосування здоровим добровольцям період напіввиведення сунітинібу та його основного активного метаболіту десетилу становить приблизно 40-60 годин та 80-110 годин відповідно.
Особливі популяції
Порушення функції печінки: Сунітініб та його основний метаболіт метаболізуються переважно печінкою. Системна експозиція після одноразової дози сунітинібу була подібною у пацієнтів з легкою або помірною печінковою недостатністю (стадії А та В за Чайлдом-П’ю) у порівнянні з пацієнтами з нормальною функцією печінки. SUTENT не вивчався у пацієнтів з тяжкою печінковою недостатністю (стадія С за Чайлдом-П’ю).
Дослідження раку виключали пацієнтів з АЛТ або АСТ> 2,5 х ГДК (верхня межа норми) або якщо ці значення були> 5,0 х ВМН через метастази в печінку.
Порушення функції нирок: Фармакокінетичний аналіз населення показав, що на видимий кліренс сунітинібу (CL / F) кліренс креатиніну не впливав у розглянутому діапазоні (42-347 мл / хв). Системна експозиція після одноразової дози сунітинібу була подібною у пацієнтів з тяжкою нирковою недостатністю ( CLcr80 мл / хв.) Хоча сунітиніб та його основний метаболіт не виводилися гемодіалізом у пацієнтів із ШОЕ, загальна системна експозиція була на 47% нижчою для сунітинібу та на 31% для його основного метаболіту порівняно з суб’єктами з нормальною функцією нирок.
Вага, стан працездатності: Фармакокінетичний аналіз населення показує, що не потрібно коригувати початкову дозу для ваги та стану працездатності Східної кооперативної онкологічної групи (ECOG).
Стать належності: Наявні дані свідчать, що у самок може бути на 30% нижчий видимий кліренс сунітинібу (CL / F), ніж у чоловіків; ця різниця, однак, не потребує корекції початкової дози.
05.3 Дані доклінічної безпеки
Дослідження токсичності повторних доз на щурах та мавпах тривалістю до 9 місяців показали, що основні ефекти на органи -мішені - у шлунково -кишковому тракті (блювота та діарея у мавп); надниркові залози (коркова скупченість та / або крововилив у щурів та мавп, з некрозом з подальшим фіброзом у щурів); кров та лімфатична система (гіпоцелюлярність кісткового мозку та виснаження лімфоїдів тимусу, селезінки та лімфатичних вузлів); екзокринна підшлункова залоза (дегрануляція ацинарних клітин з одноклітинним некрозом); слинних залоз (ацинарна гіпертрофія); кісткові суглоби (потовщення ростової пластинки); матки (атрофія) та яєчників (зменшення розвитку фолікулів). Усі результати відбулися з клінічно значущими рівнями експозиції сунітінібу в плазмі. Інші ефекти, які спостерігалися у дослідженнях, включали: подовження інтервалу QTc, зменшення фракції викиду лівого шлуночка (LVEF) та яєчка канальцева атрофія, збільшення мезангіальних клітин у нирках, крововилив у шлунково -кишковий тракт та слизову оболонку ротової порожнини та гіпертрофія клітин передньої долі гіпофіза. ендометрій) та пластинка росту кісток (потовщення епіфіза або дисплазія хряща) пов’язані з фармакологічною дією сунітинібу. Більшість цих подій були оборотними після припинення лікування протягом 2-6 тижнів.
Генотоксичність
Оцінювали генотоксичний потенціал сунітинібу in vitro та в природних умовах. Сунітініб не виявив мутагенних ефектів у бактерій, використовуючи метаболічну активацію, що забезпечується печінкою щурів. Сунітініб не викликав структурних хромосомних аберацій у периферичних лімфоцитах людини в пробірці. У периферичних лімфоцитах людини спостерігалася поліплоїдія (аберації в кількості хромосом) в vitro, як за наявності, так і за відсутності метаболічної активації. Сунітініб не виявив кластогенного потенціалу в кістковому мозку щурів в природних умовах. Основний активний метаболіт не оцінювався на генотоксичний потенціал.
Канцерогенність
У 1-місячному дослідженні для визначення діапазону доз (0, 10, 25, 75 або 200 мг / кг / добу), в якому трансгенних мишей rasH2 годували оральним зондом шляхом дозування щоденно та безперервно, при введенні спостерігалися найвищі дози (200 мг / кг / день) карциноми та гіперплазія залоз Бруннера дванадцятипалої кишки.
Було проведено 6-місячне дослідження для оцінки канцерогенності у трансгенних мишей rasH2, яких годували ротовою трубкою шляхом дозування введеної дози щодня (0, 8, 25, 75 [зменшено до 50] мг / кг / добу). При дозах ≥25 мг / кг / добу, що вводяться протягом 1 або 6 місяців (≥7,3 -кратна AUC, що спостерігається у пацієнтів, які отримують рекомендовану добову дозу [RDD]), гастродуоденальних карцином, спостерігалася підвищена частота фону. Гемангіосаркома та / або гіперплазія слизову шлунка.
У 2-річному дослідженні для оцінки канцерогенності у щурів (0, 0,33, 1 або 3 мг / кг / день), коли сунітиніб вводили 28-денними циклами з наступними 7-денними періодами без введення, було виявлено збільшення частоти феохромоцитома та медуларна гіперплазія надниркових залоз у самців щурів, які отримували дозу 3 мг / кг / добу протягом більше 1 року (≥ 7,8 разів більше AUC у пацієнтів, які отримували РДД). Карцинома залози Бруннера сталася в дванадцятипалій кишці у дозах ≥ 1 мг / кг / день у жінок, у дозах 3 мг / кг / день у чоловіків та гіперплазія клітин слизової оболонки була очевидною у залозах шлунка при рівних дозах при 3 мг / кг / добу в день у чоловіків; ці події відбувалися у дозах, що у 0,9, 7,8 та 7,8 разів перевищували AUC у пацієнтів, які отримували РДД відповідно. Доречність цих новоутворень, виявлених у дослідженнях канцерогенності мишей (rasH2) та щурів для людей, які отримували сунитиніб, невідома.
Токсичність для репродукції та розвитку
У дослідженнях репродуктивної токсичності не було виявлено жодного впливу на фертильність у чоловіків чи жінок.Однак дослідження токсичності при повторних дозах, проведені на щурах та мавпах, показали вплив на фертильність самок у вигляді фолікулярної атрезії, дегенерації жовтого тіла, змін ендометрію в матці та зменшення ваги матки та яєчників до рівня клінічно значущого. системний вплив. Вплив на фертильність самців щурів спостерігався у вигляді канальцевої атрофії в яєчках, зменшення сперматозоїдів в придатку яєчка та колоїдного виснаження в передміхуровій залозі та насіннєвих бульбашках при рівні експозиції у плазмі, що у 25 разів перевищує системну експозицію людини.
У щурів смертність ембріона та плоду призвела до значного зменшення кількості живих плодів, збільшення кількості резорбцій, збільшення втрат після імплантації та загальної втрати потомства у 8 із 28 вагітних самок при рівні експозиції у плазмі крові, що в 5,5 разів перевищує системну експозицію у людей . У кроликів зменшення ваги вагітної матки та кількості живих плодів було викликано збільшенням резорбції плоду, втратами після імплантації та загальною втратою потомства у 4 із 6 вагітних самок при 3-кратному рівні експозиції у плазмі крові. Системна експозиція у людей. Лікування сунітінібом у щурів під час органогенезу призвело до ефектів розвитку у дозах ≥ 5 мг / кг / добу, що полягало у збільшенні частоти вад розвитку скелета плода, що переважно характеризується затримкою окостеніння грудних / поперекових хребців і виникає при рівні експозиції у плазмі крові 5.5 разів більше системного впливу людини. У кроликів вплив на розвиток полягав у збільшенні частоти розщеплення губи на рівні експозиції у плазмі крові, приблизно дорівнює тій, що спостерігається у клініці, та ущелині губи та щілини на рівні експозиції у плазмі крові, що у 2,7 рази перевищує системну експозицію у людини.
Вплив сунітинібу (0,3; 1,0; 3,0 мг / кг / добу) на пре- та постнатальний розвиток оцінювали у дослідженні на вагітних самках щурів. Приріст маси тіла матері під час гестації та лактації зменшувався при дозах> 1 мг / кг / добу, але репродуктивної токсичності для матері не спостерігалося до доз 3 мг / кг / добу (розрахункова експозиція> 2, 3 -кратна AUC, виявлена у пацієнтів Зниження маси тіла нащадків спостерігалося у дозах 3 мг / кг / день у період до і після відлучення. AUC, що спостерігається у пацієнтів, які отримують RDD).
06.0 ФАРМАЦЕВТИЧНА ІНФОРМАЦІЯ
06.1 Допоміжні речовини
Вміст капсули
Маніт (E421)
Кроскармелоза натрію
Повідон (K-25)
Стеарат магнію
Оболонка капсули
Желе
Червоний оксид заліза (E172)
Діоксид титану (E171)
Чорнило
Шелак
Пропіленгліколь
Їдкий натр
Повідон
Діоксид титану (E171)
06.2 Несумісність
Не актуально.
06.3 Строк дії
3 роки.
06.4 Особливі умови зберігання
Цей лікарський засіб не вимагає особливих умов зберігання.
06.5 Характер негайної упаковки та вміст упаковки
Пляшки з поліетилену високої щільності (HDPE) з поліпропіленовою кришкою, що містять 30 твердих капсул.
Прозорі блістери з полі (хлортрифторетилену) / ПВХ з перфорованими одиничними дозами та з алюмінієвою фольгою з лаком для термоущільнення, що містить 28 х 1 твердих капсул.
Не всі розміри упаковок можна продавати.
06.6 Інструкції з використання та поводження
Ніяких спеціальних вказівок.
07.0 ВЛАСНИК РОЗРОБНИЦТВА
ТОВ "Пфайзер"
Ramsgate Road
Сендвіч, Кент CT13 9NJ
Великобританія
08.0 НОМЕР РОЗВИТКУ З РОБОТИ
ЄС/1/06/347/001
037192022
ЄС/1/06/347/004
09.0 ДАТА ПЕРШОГО ДОЗВІЛЕННЯ АБО ОНОВЛЕННЯ ДОЗВІЛЛЯ
Дата першого дозволу: 19 липня 2006 року
Дата останнього оновлення: 09 січня 2012 року
10.0 ДАТА ПЕРЕГЛЯНУ ТЕКСТУ
11.0 ДЛЯ РАДІОЛЕКЦІЙ, ПОВНІ ДАНІ ПРО ВНУТРІШНЮ РАДІАЦІЮ
12.0 ДЛЯ РАДІОЛЕКЦІЙ, ДОДАТКОВИХ ДЕТАЛЬНИХ ІНСТРУКЦІЙ З ВИКОРИСТОЇ ПІДГОТОВКИ І КОНТРОЛЮ ЯКОСТІ








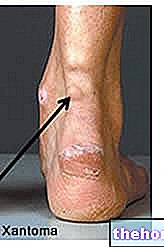











.jpg)
