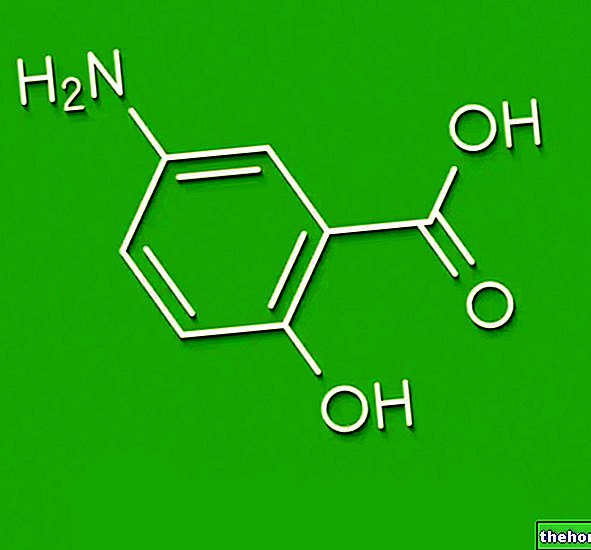
Діючі речовини: Ентекавір
Бараклюд 0,5 мг таблетки, вкриті оболонкою
Вкладиші для упаковки Baraclude доступні для розмірів упаковки:- Бараклюд 0,5 мг таблетки, вкриті оболонкою
- Бараклюд 1 мг таблетки, вкриті оболонкою
- Бараклюд 0,05 мг / мл пероральний розчин
Показання Для чого використовується Baraclude? Для чого це?
Таблетки Бараклюде-це противірусні ліки, які застосовуються у дорослих для лікування хронічної (тривалої) інфекції вірусу гепатиту В. Бараклюд можна застосовувати людям, у яких печінка пошкоджена, але все ще функціонує належним чином (компенсоване захворювання печінки), а також людям, у яких печінка пошкоджена та не функціонує належним чином (декомпенсована хвороба печінки).
Таблетки Бараклюд також використовуються для лікування хронічної (тривалої) інфекції вірусу гепатиту В у дітей та підлітків від 2 до 18 років. Baraclude можна застосовувати у дітей, у яких печінка пошкоджена, але все ще функціонує належним чином (компенсоване захворювання печінки).
Інфекція вірусом гепатиту В може викликати ураження печінки. Baraclude зменшує кількість вірусу в організмі та покращує стан печінки.
Протипоказання Коли Бараклюд не слід використовувати
Не приймайте Baraclude, якщо у вас алергія (гіперчутливість) на ентекавір або будь -який інший інгредієнт цього препарату (перерахований у розділі 6).
Заходи безпеки при застосуванні Що потрібно знати, перш ніж приймати Бараклюд
Поговоріть зі своїм лікарем або фармацевтом, перш ніж приймати Бараклюд
- якщо у вас були проблеми з нирками, повідомте про це свого лікаря. Це важливо, оскільки Baraclude виводиться з організму через нирки, і його, можливо, доведеться коригувати відповідно до дози або схеми.
- не припиняйте прийом Baraclude без порад лікаря, оскільки після припинення лікування гепатит може погіршитися. Якщо лікування Baraclude буде припинено, ваш лікар продовжуватиме спостерігати за вами та проводити аналізи крові протягом кількох місяців.
- Обговоріть зі своїм лікарем, чи працює ваша печінка належним чином, а якщо ні, то які наслідки це може вплинути на лікування Baraclude.
- якщо ви також інфіковані вірусом ВІЛ (вірус імунодефіциту людини), повідомте про це свого лікаря. Не слід приймати Baraclude для лікування інфекції гепатиту В, якщо ви вже не приймаєте ліки від ВІЛ, оскільки ефективність майбутнього лікування ВІЛ може бути знижена. Baraclude не перевірятиме його ВІЛ -інфекцію.
- Прийом Baraclude не запобігає зараженню інших людей вірусом гепатиту В (HBV) через статеві контакти або рідинами організму (включаючи зараження кров’ю). З цієї причини важливо вживати запобіжних заходів, щоб запобігти інфікуванню інших вірусом. гепатиту В (ВГВ). Доступна вакцина для захисту тих, кому загрожує зараження вірусом гепатиту В (HBV).
- Baraclude належить до класу ліків, які можуть викликати молочнокислий ацидоз (надлишок молочної кислоти в крові) та збільшення печінки. Такі симптоми, як нудота, блювота та біль у шлунку, можуть свідчити про розвиток молочнокислого ацидозу. Цей рідкісний, але серйозний побічний ефект іноді був летальним. Молочнокислий ацидоз частіше зустрічається у жінок, особливо якщо вони мають надмірну вагу. Ваш лікар буде регулярно перевіряти вас під час лікування Baraclude.
- якщо ви раніше отримували лікування хронічного гепатиту В, повідомте про це свого лікаря.
Діти та підлітки
Не слід давати Baraclude дітям до 2 років або вагою менше 10 кг.
Взаємодії Які препарати або продукти харчування можуть змінити дію Бараклюде
Повідомте свого лікаря або фармацевта, якщо ви приймаєте, нещодавно приймали або могли б приймати будь -які інші ліки.
Baraclude з їжею та напоями
У більшості випадків ви можете приймати Baraclude з їжею або без неї. Однак, якщо ви раніше отримували ліки, що містять активну речовину ламівудин, вам слід врахувати наступне. Якщо ви перейшли на лікування Baraclude через неефективність терапії ламівудином, вам потрібно буде приймати Baraclude раз на день натщесерце. Якщо захворювання печінки дуже запущене, лікар призначить вам приймати Бараклюд натщесерце. Натщесерце визначається як мінімум через 2 години після їди і принаймні за 2 години до наступного прийому їжі.
Діти та підлітки (віком від 2 до 18 років) можуть приймати Бараклюд з їжею або без неї.
Попередження Важливо знати, що:
Вагітність, грудне вигодовування та фертильність
Скажіть своєму лікарю, якщо ви вагітні або плануєте завагітніти. Не доведено, що застосування Бараклюде під час вагітності є безпечним. Якщо Вашого лікаря спеціально не вказано, Бараклюд не слід застосовувати під час вагітності. Важливо, щоб жінки дітородного віку застосовували ефективні методи під час лікування контрацептивом Бараклюд. щоб уникнути вагітності.
Не слід годувати груддю під час лікування Baraclude. Скажіть своєму лікарю, якщо ви годуєте грудьми. Невідомо, чи виділяється ентекавір, активна речовина Baraclude, у грудне молоко людини.
Водіння автомобіля та роботу з машинами
Запаморочення, втома та сонливість - поширені побічні ефекти, які можуть погіршити вашу здатність керувати автомобілем та працювати з механізмами. За будь -якими уточненнями зверніться до лікаря.
Бараклюд містить лактозу
Цей лікарський засіб містить лактозу. Якщо ваш лікар сказав вам, що у вас «непереносимість деяких цукрів, зверніться до лікаря, перш ніж приймати цей лікарський засіб.
Доза, спосіб та час введення Як користуватися Baraclude: Дозування
Не всім пацієнтам потрібно приймати однакову дозу Бараклюду.
Завжди приймайте цей препарат точно так, як вам сказав ваш лікар. Якщо є сумніви, зверніться до лікаря або фармацевта.
Для дорослих рекомендована доза становить 0,5 мг або 1 мг один раз на день перорально (перорально).
Ваша доза буде залежати від:
- якщо Ви вже проходили лікування від інфекції вірусу гепатиту В (ВГВ) та якими ліками Ви лікувалися.
- якщо у вас проблеми з нирками. Ваш лікар може призначити меншу дозу або призначити її приймати рідше одного разу на день.
- стан печінки.
Для дітей та підлітків (від 2 до 18 років) лікар вашої дитини прийме правильну дозу, виходячи з ваги вашої дитини.
Пероральний розчин Baraclude рекомендується пацієнтам з масою тіла від 10 до 32,5 кг.
Діти вагою не менше 32,6 кг можуть приймати пероральний розчин або таблетки по 0,5 мг.
Кожна доза буде вводитися один раз на день перорально (рот).
Немає рекомендацій щодо Baraclude для дітей віком до 2 років або вагою менше 10 кг.
Ваш лікар порадить вам правильну дозу. Щоб препарат був повністю ефективним і зменшив розвиток стійкості до терапії, завжди приймайте дозу, рекомендовану вашим лікарем. Приймайте Baraclude так довго, як вам скаже лікар. Ваш лікар скаже вам, коли і коли припинити лікування.
Деяким пацієнтам слід приймати Baraclude натщесерце (див. Baraclude з їжею та напоями у розділі 2). Якщо ваш лікар сказав вам приймати Бараклюд натщесерце, це означає щонайменше за 2 години після їжі та принаймні за 2 години до наступного прийому їжі.
Якщо ви забули прийняти Бараклюд
Важливо, щоб ви не пропустили жодної дози. Якщо ви пропустили дозу Бараклюду, прийміть її якомога швидше, а потім прийміть наступну дозу в потрібний час. Якщо настав час наступної дози, пропустіть пропущену дозу. Зачекайте і прийміть наступну дозу в призначений час.
Не приймайте подвійну дозу, щоб компенсувати пропущену дозу.
Не припиняйте прийом Baraclude без порад лікаря
Багато людей мають дуже важкі симптоми гепатиту, коли припиняють прийом Бараклюду. Негайно повідомте лікаря, якщо ви помітили будь -які зміни симптомів після припинення лікування.
Якщо у вас виникнуть додаткові запитання щодо застосування цього препарату, зверніться до лікаря або фармацевта.
Передозування Що робити, якщо ви прийняли занадто багато Baraclude
Якщо ви прийняли більшу кількість препарату Бараклюд, ніж слід, негайно зверніться до лікаря.
Побічні ефекти Які побічні ефекти від Baraclude
Як і всі ліки, цей препарат може викликати побічні ефекти, хоча вони виникають не у всіх.
Пацієнти, які отримували Baraclude, повідомили про такі побічні ефекти:
- поширені (принаймні 1 із 100 пацієнтів): головний біль, безсоння (нездатність спати), стомлюваність (надмірна втома), запаморочення, сонливість (сонливість), блювота, діарея, нудота, диспепсія (порушення травлення) та високий рівень печінкових ферментів кров.
- нечасті (щонайменше 1 з 1000 пацієнтів): висип (висип), випадання волосся.
- рідко (принаймні 1 з 10 000 пацієнтів): важка алергічна реакція.
Якщо у Вас виникли будь -які побічні ефекти, зверніться до лікаря або фармацевта, що включає будь -які можливі побічні ефекти, не зазначені у цій інструкції.
Повідомлення про побічні ефекти
Якщо у Вас виникли будь -які побічні ефекти, зверніться до лікаря або фармацевта, що включає будь -які можливі побічні ефекти, не зазначені у цій інструкції. Ви також можете повідомляти про побічні ефекти безпосередньо за допомогою національної системи повідомлень, наведеної у Додатку V. Повідомляючи про побічні ефекти, ви можете допомогти надати більше інформації про безпеку застосування цього лікарського засобу.
Термін придатності та утримання
Зберігайте цей препарат подалі від очей та недоступного для дітей місця.
Не використовуйте цей препарат після закінчення терміну придатності, зазначеного на флаконі, блістері або картонній упаковці після закінчення терміну придатності. Термін придатності відноситься до останнього дня місяця.
Блистерні упаковки: Не зберігати при температурі вище 30 ° C. Зберігати в оригінальній коробці.
Упаковки в пляшках: Не зберігати при температурі вище 25 ° C. Тримайте пляшку щільно закритою.
Не викидайте ліки через стічні води або побутові відходи. Попросіть свого фармацевта, як викинути ліки, які ви більше не використовуєте. Це допоможе захистити навколишнє середовище.
Дедлайн »> Інша інформація
Що містить Baraclude
- Діюча речовина - ентекавір. Кожна таблетка, вкрита плівковою оболонкою, містить 0,5 мг ентекавіру.
- Інші допоміжні речовини:
- Ядро таблетки: кросповідон, моногідрат лактози, стеарат магнію, целюлоза мікрокристалічна та повідон.
- Покриття таблеток: гіпромелоза, макрогол 400, діоксид титану (E171) та полісорбат 80 (E433).
Опис того, як виглядає Baraclude, та вміст упаковки
Таблетки, вкриті плівковою оболонкою (таблетки), мають білу або майже білу і трикутну форму. Вони позначені "BMS" з одного боку та "1611" з іншого.
Бараклюд 0,5 мг таблетки, вкриті плівковою оболонкою, випускаються у картонній упаковці, що містить 30 х 1 або 90 х 1 таблетку, вкриту плівковою оболонкою (у перфорованих одиничних блістерах із дозою) та у флаконах, що містять 30 таблеток, вкритих плівковою оболонкою.
Не всі розміри упаковок можна продавати.
Джерело з інформацією про упаковку: AIFA (Італійське агентство з лікарських засобів). Вміст, опублікований у січні 2016 р. Наявна інформація може бути не актуальною.
Щоб мати доступ до найновішої версії, бажано зайти на веб-сайт AIFA (Італійське агентство з лікарських засобів). Відмова від відповідальності та корисна інформація.
01.0 НАЗВА ЛЕКАРСТВЕННОГО ПРОДУКТУ -
БАРАКЛУД таблетки 0,5 мг, покриті плівкою
02.0 ЯКІСНИЙ І КІЛЬКІСНИЙ СКЛАД -
Кожна таблетка містить 0,5 мг ентекавіру (у вигляді моногідрату).
Допоміжні речовини з відомими ефектами: кожна таблетка містить 120,5 мг лактози
Повний список допоміжних речовин див. У розділі 6.1.
03.0 ФАРМАЦЕВТИЧНА ФОРМА -
Таблетка, вкрита плівковою оболонкою (таблетка).
Біла або брудно-біла таблетка трикутної форми з позначкою "BMS" з одного боку та "1611" з іншого.
04.0 КЛІНІЧНА ІНФОРМАЦІЯ -
04.1 Терапевтичні показання -
Бараклюд показаний для лікування хронічної інфекції вірусу гепатиту В (HBV) (див. Розділ 5.1) у дорослих з:
§ компенсоване захворювання печінки та свідчення активної реплікації вірусу, стійко підвищені рівні аланінамінотрансферази сироватки крові (АЛТ) та гістологічні дані активного запалення та / або фіброзу.
§ декомпенсована хвороба печінки (див. Розділ 4.4)
Як для компенсованої, так і для декомпенсованої хвороби печінки цей показник ґрунтується на клінічних даних у пацієнтів, які раніше не лікувалися з нуклеозидною інфекцією з HBeAg-позитивним та HBeAg-негативним вірусом гепатиту В. Для пацієнтів з резистентним до ламівудину гепатитом В див. Розділи 4.2, 4.4 та 5.1.
Бараклюд також показаний для лікування хронічної інфекції вірусу гепатиту В (HBV) у педіатричних пацієнтів, які не отримували нуклеозидів віком від 2 до 18 років з компенсованим захворюванням печінки, які мають ознаки активної реплікації вірусу та стійко підвищеного рівня аланінамінотрансферази сироватки крові (АЛТ) або гістологічні ознаки активного запалення та / або фіброзу середнього та тяжкого ступеня. Щодо рішення про початок лікування у педіатричних пацієнтів, див. розділи 4.2, 4.4 та 5.1
04.2 Дозування та спосіб введення -
Лікування повинен розпочинати лікар, який має досвід лікування хронічної інфекції вірусу гепатиту В.
Дозування
Компенсоване захворювання печінки
Пацієнти ніколи не лікувалися нуклеозидами: Рекомендована доза для дорослих становить 0,5 мг 1 раз на день, з їжею або без неї.
Пацієнти вогнетривкий до ламівудину (тобто з ознаками віремії під час лікування ламівудином або з наявністю мутацій, що викликають резистентність до ламівудину [LVDr]) (див. розділи 4.4 та 5.1): рекомендована доза для дорослих становить 1 мг 1 раз на добу, яку слід приймати натщесерце (більше ніж За 2 години до і більше ніж через 2 години після їжі) (див. Розділ 5.2). За наявності мутацій LVDr слід віддавати перевагу комбінованому застосуванню ентекавіру разом з другим противірусним засобом (який не виявляє перехресної резистентності до ламівудину або ентекавіру) перед монотерапією ентекавіром (див. Розділ 4.4).
Декомпенсоване захворювання печінки
Рекомендована доза для дорослих пацієнтів з декомпенсованою хворобою печінки - 1 мг 1 раз на добу, що приймається натщесерце (більше ніж за 2 години до та більше ніж через 2 години після їжі) (див. Розділ 5.2). Для пацієнтів з резистентним до ламівудину гепатитом В див. Розділи 4.4 та 5.1
Тривалість терапії
Оптимальна тривалість лікування невідома. Лікування можна припинити:
§ у HBeAg -позитивних дорослих пацієнтів лікування слід продовжувати щонайменше до 12 місяців після досягнення сероконверсії HBe (втрата HBeAg та негативізація ДНК HBV з появою анти -HBe у 2 послідовних вимірах сироватки, що повторюються щонайменше через 3-6 місяців) або до сероконверсії HBs або у разі втрати ефективності (див. розділ 4.4.).
§ У HBeAg негативних дорослих пацієнтів лікування слід продовжувати принаймні до сероконверсії HBs або якщо є докази втрати ефективності. При тривалому лікуванні більше 2 років рекомендується коригування, щоб підтвердити, що продовження обраної терапії залишається придатним для пацієнта.
Пацієнтам з декомпенсованою хворобою печінки або цирозом припинення лікування не рекомендується.
Педіатричне населення
Рішення про лікування педіатричних пацієнтів повинно ґрунтуватися на ретельному розгляді індивідуальних потреб пацієнта та посиланні на діючі рекомендації щодо педіатричного лікування, включаючи цінність гістологічної довідкової інформації. При продовженні терапії слід зважити переваги тривалого вірусологічного придушення проти ризику тривалого лікування, включаючи появу резистентності до вірусу гепатиту В.
Рівень аланінамінотрансферази в сироватці крові (АЛТ) слід постійно підвищувати щонайменше за 6 місяців до початку лікування у педіатричних пацієнтів з компенсованим захворюванням печінки внаслідок хронічного гепатиту В з HBeAg позитивною та протягом щонайменше 12 місяців у пацієнтів з HBeAg -негативною інфекцією. Педіатричним пацієнтам з масою тіла не менше 32,6 кг слід вводити добову дозу однієї таблетки 0,5 мг або 10 мл (0,5 мг) перорального розчину з їжею або без неї. Для пацієнтів з масою тіла менше 32,6 кг.
Тривалість терапії у педіатричних пацієнтів
Оптимальна тривалість лікування невідома. Відповідно до діючих педіатричних рекомендацій лікування можна припинити:
§ у HBeAg -позитивних педіатричних пацієнтів лікування слід продовжувати щонайменше протягом 12 місяців після зникнення ДНК HBV та сероконверсії HBeAg (втрата HBeAg та поява анти -HBe у 2 послідовних вимірах сироватки крові, що повторюються щонайменше через 3-6 місяців). до сероконверсії HBs або у разі втрати ефективності.Після припинення лікування слід регулярно контролювати рівень аланінамінотрансферази сироватки крові (АЛТ) та ДНК HBV (див. розділ 4.4).
§ У педіатричних пацієнтів з негативним HBeAg лікування слід продовжувати принаймні до сероконверсії HBsAg або якщо є докази втрати ефективності.
Фармакокінетика у педіатричних пацієнтів з нирковою або печінковою недостатністю не вивчена.
Літні громадяни: Не потрібно коригувати дозу залежно від віку. Дозу слід коригувати відповідно до функції нирок пацієнта (див. Рекомендації щодо дозування при нирковій недостатності та розділ 5.2)
Стать і раса: Не потрібно коригування статі чи раси.
Ниркова недостатність: Кліренс ентекавіру зменшується зі зменшенням кліренсу креатиніну (див. Розділ 5.2). Пацієнтам з гемодіалізом кліренсу креатиніну або на постійному амбулаторному перитонеальному діалізі (CAPD) рекомендується коригувати дозу. При застосуванні перорального розчину Бараклюде рекомендується зменшити добову дозу, як описано в таблиці. Крім того, якщо пероральний розчин недоступний, дозу можна регулювати, збільшуючи інтервал між прийомами, також описаний у таблиці. Запропоновані зміни дози засновані на екстраполяції обмежених даних, а їх безпечність та ефективність не були клінічно оцінені. Тому вірусологічну відповідь слід ретельно контролювати.
* для дозувань
** у дні гемодіалізу вводити ентекавір після гемодіалізу.
Печінкова недостатність: Не потрібно коригувати дозу для пацієнтів з печінковою недостатністю.
Спосіб введення
Baraclude слід приймати перорально.
04.3 Протипоказання -
Підвищена чутливість до активної речовини або до будь -якої з допоміжних речовин, перерахованих у розділі 6.1.
04.4 Спеціальні попередження та відповідні запобіжні заходи при використанні -
Ниркова недостатністьПацієнтам з нирковою недостатністю рекомендується коригувати дозу (див. Розділ 4.2). Запропоновані зміни дози ґрунтуються на екстраполяції обмежених даних, а відповідна безпека та ефективність не були клінічно оцінені. Тому вірусологічну реакцію необхідно ретельно контролювати.
Загострення гепатитуЗагострення, що характеризуються тимчасовим підвищенням рівня АЛТ у сироватці крові, відносно поширені при хронічному гепатиті В. Після початку противірусної терапії рівень АЛТ у сироватці крові може зростати у деяких пацієнтів, а також знижуватися рівень ДНК ВГВ (див. Розділ 4.8). Серед пацієнтів, які отримували ентекавір, середня частота спалахів під час лікування становила 4-5 тижнів. У пацієнтів із компенсованим захворюванням печінки ці підвищення рівня АЛТ у сироватці крові зазвичай не супроводжуються підвищенням концентрації білірубіну в сироватці крові або декомпенсацією печінки. Пацієнти з прогресуючим захворюванням печінки або цирозом печінки можуть бути схильні до більш високого ризику декомпенсації печінки після загострення гепатиту, тому під час терапії їх потрібно ретельно контролювати.
Повідомлялося також про гостре загострення гепатиту у пацієнтів, які припинили терапію гепатиту В (див. Розділ 4.2). Загострення після лікування, як правило, пов'язані з підвищенням рівня ДНК ВГВ, проте більшість із них, проте, спостерігалися важкі загострення, включаючи летальні випадки. .
Серед пацієнтів, які отримували ентекавір, які ніколи не отримували нуклеозиди, середня тривалість загострень після лікування становила 23-24 тижні, і більшість з них спостерігалася у пацієнтів з негативним HBeAG (див. Розділ 4.8). Функцію печінки слід контролювати періодично з клінічними та лабораторними дослідженнями щонайменше кожні 6 місяців після припинення терапії гепатитом В. При необхідності терапію гепатиту В можна відновити.
Пацієнти з декомпенсованою хворобою печінки: "Високий рівень серйозних побічних ефектів з боку печінки (незалежно від причинно-наслідкових зв'язків) спостерігався у пацієнтів з декомпенсованою хворобою печінки, особливо у пацієнтів з хворобою Чайлда-Туркотта-П'ю (CTP) класу С, порівняно з відсотками, виявленими у пацієнтів з компенсованою функцією печінки Крім того, пацієнти з декомпенсованою хворобою печінки можуть мати вищий ризик розвитку молочнокислого ацидозу та специфічних побічних ефектів з боку нирок, таких як гепаторенальний синдром. Тому у цій популяції пацієнтів слід ретельно контролювати клінічні та лабораторні параметри (див. Також розділи 4.8 та 5.1) .
Молочнокислий ацидоз та важка гепатомегалія зі стеатозом: при застосуванні нуклеозидних аналогів повідомлялося про молочнокислий ацидоз (за відсутності гіпоксемії), іноді смертельний, зазвичай пов’язаний з важкою гепатомегалією та стеатозом печінки. Оскільки ентекавір є аналогом нуклеозидів, цей ризик не можна виключати. Лікування аналогами нуклеозидів слід припинити у разі підвищення рівня амінотрансферази, прогресуючої гепатомегалії або метаболічного / молочнокислого ацидозу невідомої етіології. , були пов'язані з панкреатитом, печінковою недостатністю / жировою хворобою печінки, нирковою недостатністю та підвищенням рівня молочної кислоти в сироватці крові. Необхідно бути обережним при введенні аналогів нуклеозидів пацієнтам (особливо жінкам з ожирінням) з гепатомегалією, гепатитом або іншими відомими факторами ризику захворювання печінки За цими пацієнтами слід уважно стежити розум.
Щоб диференціювати підвищення амінотрансфераз внаслідок відповіді на лікування від тих, які потенційно пов’язані з молочнокислим ацидозом, лікарі повинні переконатися, що зміни АЛТ пов’язані зі збільшенням інших лабораторних маркерів хронічного гепатиту В.
Стійкість та особливі запобіжні заходи для пацієнтів вогнетривкий до ламівудину: Мутації в полімеразі HBV, які декодують заміни на резистентність до ламівудину, можуть призвести до подальшого виникнення вторинних заміщень, включаючи ті, що пов'язані з резистентністю до ентекавіру (ETVr). У невеликому відсотку пацієнтів, стійких до ламівудину, мутації ETVr до rtT184, rtS202 або rtM & SUP2; 50 були присутніми на початковому етапі. Пацієнти з резистентним до ламівудину гепатитом С мають підвищений ризик розвитку подальшої резистентності до ентекавіру порівняно з пацієнтами, не резистентними до ламівудину. 4 та 5-річний курс лікування у пацієнтів з резистентними до ламівудину пацієнтами становив відповідно 6%, 15%, 36%, 47%та 51%. Вірусологічну відповідь слід часто контролювати у вогнетривкій популяції. Ламівудин та відповідне тестування на резистентність слід проводити пацієнти з субоптимальною вірусологічною відповіддю після 24 серій Під час лікування ентекавіром слід розглянути можливість корекції лікування (див. Розділи 4.5 та 5.1). На початку терапії у пацієнтів з документально підтвердженою історією резистентності до ламівудину ВГВ слід віддавати перевагу комбінованому застосуванню ентекавіру та другого противірусного засобу (який не виявляє перехресної резистентності до ламівудину або ентекавіру) перед монотерапією ентекавіром. пов'язаний з підвищеним ризиком подальшої резистентності до ентекавіру незалежно від ступеня захворювання печінки; вірусологічний прорив може бути пов'язаний з важкими клінічними ускладненнями основного захворювання печінки у пацієнтів з декомпенсованою хворобою печінки. слід віддавати перевагу комбінованому застосуванню ентекавіру плюс другого противірусного засобу (який не виявляє перехресної резистентності до ламівудину або ентекавіру) перед монотерапією ентекавіром.
Педіатричне населення: Нижчий рівень вірусологічної відповіді (ДНК HBV
Трансплантація печінки: Перед та під час терапії ентекавіром у пацієнтів, які отримували трансплантат печінки, які отримували циклоспорин або такролімус, слід ретельно оцінити функцію нирок (див. Розділ 5.2).
Спільна інфекція з гепатитом С або D: Немає даних про ефективність ентекавіру у пацієнтів, які одночасно інфіковані вірусами гепатиту С або D.
Пацієнти з вірусом імунодефіциту людини (ВІЛ) / ВГВ, які не отримують одночасно антиретровірусну терапію: ентекавір не оцінювався у пацієнтів, коінфікованих ВІЛ / ВГВ, які не отримували супутнього ефективного лікування ВІЛ.Початок резистентності до ВІЛ спостерігався, коли ентекавір застосовувався для лікування хронічної інфекції гепатиту В. HAART) (див. Розділ 5.1). Тому терапію ентекавіром не слід застосовувати для пацієнтів, коінфікованих ВІЛ / ВГВ, які не отримують лікування ВААРТ. Ентекавір не вивчався для лікування ВІЛ -інфекції і не рекомендується для цього застосування.
Пацієнти, коінфіковані ВІЛ / ВГВ, які одночасно отримують антиретровірусну терапію: ентекавір вивчали у 68 дорослих, коінфікованих ВІЛ / ВГВ, які отримували ВААРТ, що містить ламівудин (див. розділ 5.1). Немає даних про ефективність ентекавіру у ВІЛ-інфікованих HBeAg-негативних пацієнтів. Існують обмежені дані про пацієнтів, коінфікованих ВІЛ, з низькою кількістю клітин CD4 (клітин / мм3).
Загальні: Пацієнтів слід попередити, що терапія ентекавіром не знижує ризик передачі гепатиту В, тому слід продовжувати вживати відповідних запобіжних заходів.
Лактоза: Кожна добова доза цього лікарського засобу 0,5 мг містить 120,5 мг лактози.
Пацієнти з рідкісними спадковими проблемами непереносимості галактози, дефіцитом лактази Лаппа або мальабсорбцією глюкози-галактози не повинні приймати цей препарат. Для цих осіб доступний пероральний розчин Baraclude, який не містить лактози.
04.5 Взаємодія з іншими лікарськими засобами та інші форми взаємодії -
Оскільки ентекавір виводиться переважно нирками (див. Розділ 5.2), одночасне застосування з лікарськими засобами, що знижують функцію нирок або конкурують з активною канальцевою секрецією, може збільшити концентрацію обох лікарських засобів у сироватці крові. Окрім ламівудину, адефовіру дипівоксилу та тенофовіру дизопроксилу фумарату, ефекти одночасного застосування ентекавіру з лікарськими засобами, які виводяться нирками або впливають на функцію нирок, не оцінювалися. За пацієнтами слід ретельно контролювати наявність небажаних ефектів, які можуть виникнути під час одночасного застосування ентекавіру з такими лікарськими засобами.
Фармакокінетичних взаємодій між ентекавіром та ламівудином, адефовіром та тенофовіром не спостерігалося.
Ентекавір не є субстратом, індуктором або інгібітором ферментів цитохрому Р450 (CYP450) (див. Розділ 5.2). Тому малоймовірно, що з ентекавіром відбудеться лікарська взаємодія, що здійснюється через CYP450.
Педіатричне населення
Дослідження взаємодії проводилися тільки у дорослих.
04.6 Вагітність та годування груддю -
Жінки дітородного віку: Оскільки потенційні ризики для розвитку плода невідомі, жінкам репродуктивного віку слід застосовувати ефективні засоби контрацепції.
Вагітність: Не існує адекватних досліджень щодо застосування ентекавіру у вагітних жінок.Дослідження на тваринах показали репродуктивну токсичність у високих дозах (див. Розділ 5.3).
Потенційний ризик для людини невідомий. Baraclude не слід застосовувати під час вагітності, якщо це не є крайньою необхідністю. Немає даних про вплив ентекавіру на передачу ВГВ від матері до новонародженого. Тому слід вжити відповідних заходів для запобігання зараженню ВГВ у новонароджених.
Час годування: невідомо, чи виділяється ентекавір у жіноче молоко. Наявні токсикологічні дані у тварин показали екскрецію ентекавіру у грудне молоко (докладніше див. Розділ 5.3). Не можна виключити ризик для дітей. Під час терапії Бараклюдом годування груддю слід припинити.
Родючість: Токсикологічні дослідження на тваринах, яким вводили ентекавір, не показали жодних ознак втрати фертильності (див. Розділ 5.3).
04.7 Вплив на здатність керувати автомобілем та працювати з механізмами -
Досліджень щодо здатності керувати транспортними засобами та працювати з механізмами не проводилося.
04.8 Побічні ефекти -
до Короткий опис профілю безпеки
У клінічних дослідженнях пацієнтів з компенсованою хворобою печінки найчастішими побічними реакціями будь -якого ступеня тяжкості, принаймні одним можливим зв’язком з ентекавіром, були: головний біль (9%), втома (6%), запаморочення (4%) та нудота ( 3%). Повідомлялося про загострення гепатиту під час та після припинення терапії ентекавіром (див. Розділ 4.4 c. Опис окремих побічних реакцій).
b. Перелік побічних реакцій
Оцінка побічних реакцій ґрунтується на досвіді постмаркетингового спостереження та на чотирьох клінічних дослідженнях, в яких 1720 пацієнтів з хронічною інфекцією вірусу гепатиту В та компенсованою хворобою печінки лікувалися подвійним сліпим способом ентекавіром (n = 862) або ламівудином. (N = 858) до 107 тижнів (див. Розділ 5.1). У цих дослідженнях профілі безпеки, включаючи зміни лабораторних показників, були подібними для ентекавіру 0,5 мг один раз на день (679 пацієнтів, які раніше не отримували нуклеозидний HBeAg, або негативних протягом середньої межі 53 тижнів), ентекавіру 1 мг один раз на день (183 ламівудину, стійкого до впливу ламівудину) пацієнти, які лікувалися протягом середини 69 тижнів) та ламівудин.
Побічні реакції, що вважаються принаймні можливо пов'язаними з лікуванням ентекавіром, перераховані за класами системних органів. Частота визначається як дуже часта (≥ 1/10); поширені (≥ 1/100 до
Порушення імунної системи: рідко: анафілактоїдна реакція
Психічні розлади: поширені: безсоння
Розлади нервової системи: поширені: головний біль, запаморочення, сонливість
Шлунково -кишкові розлади: часто: блювота, діарея, нудота, диспепсія
Гепатобіліарні порушення поширені: підвищення рівня трансаміназ
Порушення з боку шкіри та підшкірної клітковини: нечасто: висип, алопеція
Загальні розлади та стан на місці введення: поширені: втома
Повідомлялося про випадки молочнокислого ацидозу, часто пов'язаного з декомпенсацією печінки, іншими серйозними захворюваннями або впливом препарату (див. Розділ 4.4).
Лікування понад 48 тижнів: Продовження лікування ентекавіром із середньою тривалістю 96 тижнів не виявило нових сигналів безпеки.
c. Опис окремих побічних реакцій
Аномалії лабораторних досліджень: У клінічних випробуваннях у пацієнтів, які раніше не отримували нуклеозиди, у 5% спостерігалося підвищення АЛТ> в 3 рази від вихідного рівня та у 2 рази у пацієнтів від вихідного рівня разом із загальним білірубіном> у 2 рази вище норми (верхня межа норми, ULN ) і> 2 рази в порівнянні з базовими значеннями. Рівень альбуміну амілази> 3 рази від базового рівня у 2%, рівень ліпази> 3 рази від базового рівня у 11% та тромбоцитів
У клінічних випробуваннях у пацієнтів з резистентною терапією ламівудином у 4% спостерігалося підвищення АЛТ> 3-кратного вихідного рівня та 2-кратного відсотка вихідного рівня разом із загальним білірубіном> 2-кратною ВГН та> 2 рази порівняно з вихідними значеннями. Рівень амілази> 3 рази від базового рівня спостерігався у 2% пацієнтів, рівень ліпази> 3 рази від базового рівня у 18% та тромбоцитів
Спалахи під час лікування: У дослідженнях пацієнтів, які раніше не отримували нуклеозиди, під час лікування у 2 % пацієнтів, які отримували ентекавір проти 4 % пацієнтів, які отримували ламівудин, спостерігалося підвищення рівня АЛТ> 10 разів вище ГМН та> 2-кратного вихідного рівня. У дослідженнях пацієнтів з резистентною терапією ламівудином під час лікування у 2% пацієнтів, які отримували ентекавір, порівняно з 11% пацієнтів, які отримували ламівудин, спостерігалося підвищення рівня АЛТ> 10 разів вище верхньої межі норми та> 2 рази порівняно з вихідним показником. середній час до підвищення від 4 до 5 тижнів, як правило, припинявся при продовженні лікування і в більшості випадків був пов'язаний із зниженням вірусного навантаження ≥ 2 log10 / мл, що передувало або збігалося з підвищенням рівня АЛТ. Під час лікування рекомендується періодичний контроль функції печінки.
Загострення після припинення лікування: у пацієнтів, які припинили лікування вірусу гепатиту В, включаючи терапію ентекавіром (див. Розділ 4.4), повідомлялося про гострі загострення гепатиту. Підвищення рівня АЛТ (> 10 разів вище ВМН та> 2 рази порівняно з референтними значеннями [найнижчі значення на початковому рівні або вимірювання при «останній введеній дозі») під час спостереження після лікування. Серед пацієнтів, які раніше не отримували нуклеозиди, які отримували ентекавір, підвищення АЛТ в середньому тривало до 23–24 тижнів, а 86% (24/28) підвищення АЛТ спостерігалося у пацієнтів з негативним HBeAg. обмежена кількість пацієнтів проходила спостереження, у 11% пацієнтів, які отримували лікування ентекавіром, і у жодного з пацієнтів, які отримували ламівудин, не спостерігалося підвищення рівня АЛТ під час спостереження після лікування.
У клінічних випробуваннях лікування ентекавіром припиняли, якщо у пацієнтів була досягнута специфічна відповідь. Якщо лікування припиняється незалежно від відповіді на терапію, частота підвищення АЛТ після лікування може бути вищою.
d. Педіатричне населення
Безпека застосування ентекавіру у педіатричних пацієнтів віком від 2 до 8 років ґрунтується на двох поточних клінічних випробуваннях на хронічно інфікованих суб’єктах з ВГВ; Фаза 2 фармакокінетичного дослідження (дослідження 028) та Фаза 3 дослідження (дослідження 189). У цих дослідженнях були задіяні 173 суб’єкти HBeAg, які ніколи раніше не лікувалися нуклеозидами та лікувалися ентекавіром протягом середньої тривалості 60 тижнів. Побічні реакції, які спостерігалися у педіатричних суб’єктів, які отримували лікування ентекавіром, відповідали тим, що спостерігалися у клінічних дослідженнях ентекавіру у дорослих (див. Короткий опис профілю безпеки та розділ 5.1).
І. Інші особливі популяції
Досвід у пацієнтів з декомпенсованою хворобою печінки: Профіль безпеки ентекавіру у пацієнтів з декомпенсованою хворобою печінки оцінювали у рандомізованому відкритому порівняльному дослідженні, в якому пацієнти отримували 1 мг ентекавіру на добу (n = 102) або адефовір дипівоксил 10 мг / добу (n = 89) (дослідження 048). Порівняно з побічними реакціями, зазначеними у розділі b. Перелік побічних реакцій, «додаткова побічна реакція [зниження бікарбонату крові (2%)] спостерігалася у пацієнтів, які отримували ентекавір до 48 тижня. Сукупна смертність за час дослідження становила 23% (23/102), а причини смерть, як правило, була пов'язана з печінкою, як і очікувалося у цій популяції. Кумулятивна частота гепатоцелюлярної карциноми (ГЦК) за час дослідження становила 12% (12/102). 69% за час дослідження Пацієнти з високим показником CTP на початковому етапі мали підвищений ризик розвитку серйозних побічних явищ (див. розділ 4.4).
Зміни лабораторних досліджень: Серед пацієнтів з декомпенсованою хворобою печінки, які отримували лікування ентекавіром до 48 тижня, жоден з них не мав підвищення АЛТ> 10 разів верхньої межі норми (ВМН) і> 2 рази вихідного рівня, ел »1% пацієнтів мали підвищення АЛТ> 2 рази вихідний рівень разом із загальним білірубіном> у 2 рази вище верхньої межі норми (ГМН) та> 2 рази від базового рівня. рівень альбуміну в 3 рази від базового рівня.
Досвід у пацієнтів, коінфікованих ВІЛ: Профіль безпеки ентекавіру у обмеженої кількості пацієнтів, коінфікованих ВІЛ / ВГВ, які проходили лікування ВААРТ (високоактивна антиретровірусна терапія), був подібний до профілю безпеки пацієнтів з моноінфікованою інфекцією з ВГВ (див. Розділ 4.4). ).
Стать / вік: Явних відмінностей у профілі безпеки ентекавіру щодо статі (≈ 25% жінок у клінічних випробуваннях) чи віку (≈ 5% пацієнтів> 65 років) не було.
Повідомлення про підозрювані побічні реакції
Повідомлення про підозрювані побічні реакції, що виникають після реєстрації лікарського засобу, є важливими, оскільки вони дозволяють постійно контролювати співвідношення користі / ризику лікарського засобу. Медичних працівників просять повідомляти про будь -які підозрювані побічні реакції через національну систему звітності.
04.9 Передозування -
Повідомляється про обмежену кількість випадків передозування ентекавіру у пацієнтів. У здорових осіб, які отримували до 20 мг / добу протягом 14 днів та разові дози до 40 мг, не було несподіваних побічних реакцій. У разі передозування пацієнта слід контролювати на наявність ознак токсичності та призначити відповідне стандартне підтримуюче лікування.
05.0 ФАРМАКОЛОГІЧНІ ВЛАСТИВОСТІ -
05.1 "Фармакодинамічні властивості -
Фармакотерапевтична група: противірусні засоби для системного застосування, нуклеозиди та нуклеотиди, інгібітори зворотної транскриптази
Код ATC: J05AF10
Механізм дії: ентекавір, нуклеозидний аналог гуанозину, активний щодо полімерази HBV, ефективно фосфорилюється в активну трифосфатну (ТП) форму, яка має внутрішньоклітинний період напіврозпаду 15 годин. функціонально він пригнічує 3 активності вірусної полімерази: праймування полімерази HBV, зворотну транскрипцію негативної нитки ДНК, починаючи з прегемонічної РНК -месенджера та синтез позитивної нитки ДНК HBV. Кі ентекавіру-ТР для ДНК-полімерази HBV становить 0,0012 мкМ. Ентекавір-ТП є слабким інгібітором клітинної ДНК-полімерази α, β та δ зі значеннями Ki від 18 до 40 мкМ. Крім того, висока експозиція ентекавіру не має значних значень несприятливий вплив на γ -полімерази або синтез ДНК мітохондрій на клітинах HepG2 (Ki> 160 мкМ).
Противірусна активність: ентекавір інгібував синтез ДНК HBV (зниження на 50%, EC50) у концентрації 0,004 мкМ у клітинах HepG2 людини, переінфікованих HBV дикого типу. Середнє значення EC50 для ентекавіру проти LVDr HBV (rtL 180M та rtM204V) становило 0,026 мкМ (діапазон 0,010 - 0,059 мкМ) Рекомбінантні віруси з адефовір -стійкими замінами rtN236T або rtA181V залишалися повністю сприйнятливими до ентекавіру.
Аналіз інгібуючої активності ентекавіру щодо лабораторної панелі та клінічних ізолятів ВІЛ-1, проведених з використанням різних клітин та методів, дозволив отримати значення EC50 у діапазоні від 0,026 до> 10 мкМ; найнижчі значення EC50 спостерігалися під час тесту Низькі рівні У клітинній культурі ентекавір відбирав заміну M184I у мікромолярних концентраціях, що підтверджує інгібуючий тиск ентекавіру у високих концентраціях. Варіанти ВІЛ, що містять заміну M184V, продемонстрували втрату сприйнятливості до ентекавіру (див. розділ 4.4).
У комбінованому аналізі HBV в культурі клітин, абакавір, диданозин, ламівудин, ставудин, тенофовір або зидовудин не були антагоністами активності ентекавіру проти ВГВ у великому відсотку концентрацій. У противірусному аналізі на ВІЛ ентекавір у мікромолярних концентраціях не був антагоністом проти активності проти ВІЛ в культурі клітин з цих шести НІЗТ або емтрицитабіну.
Стійкість у культурі клітин: щодо HBV дикого типу, віруси LVDr, що містять заміни rtM204V та rtL180M у зворотній транскриптазі, показують 8-кратне зниження сприйнятливості до ентекавіру. викликає зниження сприйнятливості до ентекавіру в культурі клітин. Заміни, що спостерігаються у клінічних ізолятах (rtT184A, C, F, G, I, L, M або S; rtS202 C, G або I; та / або rtM250I, L або V), призводять до додаткове зменшення сприйнятливості до ентекавіру у 16-741 рази порівняно з вірусом дикого типу. Одиночні заміни ETVr (стійкість до ентекавіру) rtT184, rtS202 та rtM250 мають лише помірний вплив на сприйнятливість до ентекавіру і не спостерігалися за відсутності заміщення LVDr. (резистентність до ламівудину) у більш ніж 1000 зразках пацієнтів. ерза ВГВ та резистентний ВГВ демонструють знижену здатність до реплікації в культурі клітин.
Клінічний досвід: Демонстрація користі базується на гістологічних, вірусологічних, біохімічних та серологічних реакціях після 48 тижнів лікування в активних контрольованих клінічних випробуваннях 1633 дорослих з хронічною інфекцією гепатиту В, доказами реплікації вірусу та компенсованого захворювання печінки. Безпеку та ефективність ентекавіру також оцінювали у контрольованому клінічному дослідженні у 191 пацієнта, інфікованого ВГВ, з декомпенсованою хворобою печінки та у клінічному дослідженні у 68 пацієнтів, коінфікованих ВГВ та ВІЛ.
У дослідженнях у пацієнтів з компенсованою хворобою печінки гістологічне поліпшення визначалося як ≥ 2-бальне зниження некро-запального індексу Knodell від вихідного рівня без погіршення показників фіброзу Knodell. Відповіді для пацієнтів з одним вихідним показником фіброзу Knodell 4 (цироз) були порівнянні з всі відповіді на всі заходи ефективності (у всіх пацієнтів було компенсоване захворювання печінки). Високі показники показників гістологічної активності Knodell (HAI) (> 10) були пов'язані з більшим гістологічним поліпшенням у пацієнтів, які раніше не отримували нуклеозиди. 9,0 log 10 копій / мл були пов'язані з високими показниками вірусологічної відповіді (тиждень 48 ДНК HBV
Досвід у пацієнтів, які раніше не отримували нуклеозиди, з компенсованою хворобою печінкиРезультати 48-тижневих рандомізованих подвійних сліпих досліджень, що порівнювали ентекавір (ETV) та ламівудин (LVD) у HBeAg-позитивних та HBeAg-негативних пацієнтів, наведені в таблиці нижче:
* значення р проти ламівудину
пацієнти з оцінюваною гістологією на початковому етапі (вихідний некрозапальний бал за вузлом Кноделла ≥ 2)
б основна мета
ПЛР -аналіз cRoche Cobas Amplicor (LLOQ = 300 копій / мл)
Досвід у пацієнтів вогнетривкий до ламівудину з компенсованою хворобою печінки: У подвійному сліпому рандомізованому дослідженні ламівудин-рефрактерних HBeAg-позитивних пацієнтів, у яких 85% пацієнтів з мутаціями LVDr на початковому етапі, пацієнти, які приймали ламівудин на початку дослідження, були переведені на ентекавір 1 мг один раз на день, без періоду вимивання або періоду перекриття ( n = 141) або продовження прийому 100 мг ламівудину один раз на день (n = 145). Результати через 48 тижнів наведені в таблиці нижче.
* значення р проти ламівудину
пацієнти з оцінюваною гістологією на початковому етапі (вихідний некрозапальний бал за вузлом Кноделла ≥ 2)
б основна мета
ПЛР -аналіз cRoche Cobas Amplicor (LLOQ = 300 копій / мл)
Результат після 48 тижнів лікування:
Лікування припиняли, коли на 48 тижні або протягом другого року терапії були досягнуті критерії відповіді. Критерієм відповіді було вірусологічне придушення HBV (ДНК HBV
Пацієнти ніколи не лікувалися нуклеозидами:
HBeAg позитивний (дослідження 022): лікування ентекавіром протягом 96 тижнів (n = 354) призвело до кумулятивної частоти відповіді 80% для ДНК ВГВ
Наприкінці дозування серед пацієнтів, які продовжували лікування більше 52 тижнів (середня 96 тижнів), 81% з 243 пацієнтів, які отримували ентекавір, та 39% із 164 пацієнтів, які отримували ламівудин, мали ДНК ВГВ.
HBeAg негативний (дослідження 027): Лікування ентекавіром до 96 тижнів (n = 325) призвело до сукупної частоти відповіді 94% на ДНК HBV
Для 26 пацієнтів, які отримували ентекавір та 28 пацієнтів, які отримували ламівудин, які продовжували лікування більше 52 тижнів (медіана 96 тижнів), 96% пацієнтів, які отримували ентекавір, та 64% пацієнтів, які отримували ламівудин, наприкінці дозування мали ДНК ВГВ
Для пацієнтів, які відповіли на протокол, критерій відповіді зберігався протягом 24 тижнів спостереження після лікування у 75 % (83/111) з тих, хто відповів на лікування ентекавіром, до 73 % (68/93) з них які відповіли на лікування ламівудином у дослідженні 022 та 46% (131/286) тих, хто відповів на ентекавір проти 31% (79/253) тих, хто відповів на лікування ламівудином у дослідженні 027. Протягом 48 тижнів після спостереження після Наприкінці лікування значна кількість HBeAg -негативних пацієнтів втратила відповідь.
Результати біопсії печінки: 57 пацієнтів з ключових досліджень, проведених у пацієнтів, які не отримували нуклеозидів 022 (позитивний HBeAg) та 027 (негативний HBeAg), були включені до дослідження перекидання довгостроково, їх оцінювали за довгостроковими гістологічними результатами печінки. У ключових дослідженнях доза ентекавіру становила 0,5 мг на день (середня експозиція 85 тижнів), а в основному дослідженні перекидання 1 мг на добу (середня експозиція 177 тижнів) і спочатку 51 пацієнт у перекидання вони також отримували ламівудин (середня тривалість 29 тижнів). З цих пацієнтів 55/57 (96%) мали гістологічне поліпшення, як визначено вище (див. Вище), а 50/57 (88%) знизили показник фіброзу Ішака ≥ 1 бал. Серед пацієнтів з показником фіброзу Ішака ≥ 2 на початку, 25/43 (58%) показали зниження ≥ 2 балів. Усі пацієнти (10/10) з вихідним фіброзом або прогресуючим цирозом (бал фіброзу Ішака 4, 5 або 6) мали зниження на ≥ 1 бал (середнє зниження від вихідного рівня становило 1,5 бала). На момент тривалої біопсії всі пацієнти мали ДНК HBV
Вогнетривки до ламівудину:
HBeAg позитивний (дослідження 026): Лікування ентекавіром протягом 96 тижнів (n = 141) призвело до 30% сукупної швидкості відповіді на перетворення HBeAg у сироватці крові ДНК HBV.
У 77 пацієнтів, які продовжували лікування ентекавіром після 52 тижнів (медіана 96 тижнів), 40% пацієнтів зазнали ДНК ВГВ
Вік / стать:
Не було виявлено явної різниці в ефективності ентекавіру щодо різниці між статтю (≈ 25% жінок у клінічних дослідженнях) або віком (≈ 5% пацієнтів> 65 років).
Особливі популяції
Пацієнти з декомпенсованою хворобою печінки: У дослідженні 048 191 пацієнт з HBeAg -позитивною або негативною хронічною інфекцією HBV та ознаками декомпенсації печінки, визначеної як показник CTP 7 або вище, отримували ентекавір 1 мг один раз на день або адефовір дипівоксил 10 мг один раз на день. Пацієнти ніколи не отримували лікування ВГВ або вже проходили попереднє лікування (виключаючи попереднє лікування ентекавіром, адефовіром дипівоксилом або тенофовіру дизопроксилу фумаратом). На початковому етапі пацієнти мали середній бал CTP 8,59, і 26% пацієнтів були CTP класу C. Середній показник моделі кінцевої стадії печінкової хвороби (MELD) на початковому етапі становив 16,23. Середня концентрація ДНК HBV у сироватці крові за допомогою ПЛР становила 7,83 log10 копій / мл, а середня концентрація АЛТ у сироватці крові становила 100 Од / л; 54% пацієнтів були позитивними до HBeAg, а у 35% пацієнтів заміни LVDr на Ентекавір були вищими за адефовір дипівоксил у первинна кінцева точка ефективності, що оцінює середні зміни від вихідного рівня концентрації ДНК HBV у сироватці крові за допомогою ПЛР на 24 тижні. Результати вибраних кінцевих точок дослідження на 24 та 48 тижнях наведені в табл.
ПЛР -аналіз з амплікором Roche COBAS (LLOQ = 300 копій / мл).
bNC = F (пацієнт, який не завершив = невдача), означає, що лікування було припинено до тижня аналізу, включаючи такі причини, як смерть, відсутність ефективності, побічні явища, відсутність прихильності / втрати спостереження, що вважається невдачею (наприклад, ДНК HBV ≥ 300 копій / мл)
cNC = M (пацієнти не завершені = відсутні)
d Визначається як зменшення або відсутність зміни базової оцінки балів CTP
Початкова оцінка eMeD MELD становила 17,1 для ETV та 15,3 для адефовіру дипівоксилу.
f Знаменник - це кількість пацієнтів з аномальними значеннями на початку.
* стор
ULN = верхня межа норми, LLN = нижня межа норми.
Час до початку ГЦК або смерті (залежно від того, що сталося раніше) був порівнянним у двох групах лікування; сукупний рівень смертності за час дослідження становив 23% (23/102) та 33% (29/89) для пацієнтів, які отримували ентекавір та адефовір дипівоксил відповідно, а сукупний рівень ГЦК за час дослідження становив 12% ( 12/102) та 20% (18/89) для ентекавіру та адефовіру дипівоксилу відповідно.
Для пацієнтів з вихідними замінами LVDr відсоток пацієнтів з ДНК HBV
Пацієнти, коінфіковані ВІЛ / ВГВ, які одночасно отримували ВААРТ: Дослідження 038 включало 67 HBeAg позитивних та 1 HBeAg негативних пацієнтів, коінфікованих ВІЛ. У пацієнтів протягом 24 тижнів спостерігався стабільний, контрольований ВІЛ (плацебо з РНК ВІЛ (n = 17), потім ще 24 тижні, коли всім пацієнтам давали ентекавір. Через 24 тижні зниження вірусного навантаження на ВГВ було значно більшим. З ентекавіром (-3,65) порівняно зі збільшенням на 0,11 log10 копій / мл) .У пацієнтів, які спочатку були призначені на лікування ентекавіром, зниження ДНК HBV на 48 тижні становило -4,20 log10 копій / мл, нормалізація АЛТ виявилася у 37% пацієнтів з вихідними відхиленнями АЛТ і жодної досягнуто сероконверсії HBeAg.
Пацієнти, коінфіковані ВІЛ / ВГВ, які не супроводжуються терапією ВААРТ: ентекавір не оцінювався у пацієнтів, коінфікованих ВІЛ / ВГВ, які не отримували супутнє ефективне лікування ВІЛ.Повідомлялося про зниження РНК ВІЛ у пацієнтів, коінфікованих ВІЛ / ВГВ, які отримували монотерапію ентекавіром без ВААРТ. У деяких випадках спостерігався вибір варіанта ВІЛ -інфекції M184V, що може вплинути на вибір схем ВААРТ, які пацієнт може прийняти в майбутньому. Тому ентекавір не слід застосовувати у цьому типі населення через потенційний розвиток резистентність до ВІЛ (див. розділ 4.4).
Трансплантація печінки: Безпеку та ефективність ентекавіру 1 мг 1 раз на добу оцінювали в одноразовому дослідженні у 65 пацієнтів, яким проводили трансплантацію печінки з приводу ускладнень хронічної інфекції ВГВ, і виявляли кавказьку та 37% азіатську ДНК ВГВ із середнім віком 49 років: 89% На момент трансплантації пацієнти були HBeAg-негативними. З 61 пацієнта, який оцінювався на ефективність (вони отримували ентекавір протягом щонайменше 1 місяця), 60 також отримували імуноглобулін проти гепатиту В (HBIg) у рамках схеми профілактики після трансплантації. ці 60 пацієнтів, 49 отримували терапію HBIg протягом більше 6 місяців. На 72-му тижні після трансплантації жоден із 55 випадків, що спостерігалися, не показав реактивації вірусної реплікації [визначено як ДНК HBV ≥ 50 МО / мл (приблизно 300 копій / мл)] , а у решти 6 виключених пацієнтів вірусологічної реактивації HBV не повідомлялося. Усі 61 пацієнти продемонстрували втрату HBsAg після трансплантації, а 2 з них виявили позитивний HBsAg, зберігаючи невизначений рівень ДНК HBV (
Педіатричне населення: Дослідження 189 - це триваюче дослідження, яке базується на ефективності та безпеці ентекавіру, включаючи 180 дітей та підлітків, віком від 2 до 18 років, які раніше не лікувалися нуклеозидами та страждали на хронічний гепатит В з ураженням HBeAg, компенсованим захворюванням печінки та підвищеним вмістом АЛТ Суб'єкти були рандомізовані (2: 1) для одержання сліпого лікування ентекавіром від 0,015 мг / кг до 0,5 мг / день (N = 120) або плацебо. (N = 60). Рандомізація була стратифікована за віковою групою (від 2 до 6 років) ;> Від 6 до 12 років; і> від 12 до
24% (20/82) суб'єктів у групі ентекавіру та 2% (1/41) суб'єктів у групі плацебо досягли основної кінцевої точки. На 48 тижні 46% (38/82) суб'єктів, які отримували ентекавір та 2% (1/41) суб’єктів, які отримували плацебо, досягли значень ДНК HBV
У 2 педіатричних дослідженнях (дослідження 028 та 189) 110 пацієнтів, які отримували ентекавір протягом 48 тижнів, відстежували на резистентність. Генотипні оцінки були проведені у всіх пацієнтів, які мали вірусологічний прорив або ДНК HBV ≥ 50 МО / мл на 48 тижні або які рано припинили терапію. Заміни амінокислот, пов'язані з резистентністю до ентекавіру, не виявлено.
Клінічна стійкість: Пацієнти, які спочатку отримували в ході клінічних випробувань ентекавір 0,5 мг (без нуклеозидів) або 1,0 мг (ламівудин, стійкий до ламівудину), а також 24-тижневе вимірювання ДНК ВГВ за допомогою ПЛР під час терапії або після цього, за ними проводився моніторинг резистентності. дослідження до 240 тижнів у пацієнтів, які не отримували нуклеозидів, генотипові докази заміни ETVr у rtT184, rtS202 або rtM250 були виявлені у 3 пацієнтів, які отримували ентекавір. 2 з них також мали вірусологічний прорив (див. таблицю). Ці заміни спостерігалися лише у присутності замін LVDr (rtM204V та rtL180M).
a Результати відображають використання дози 1 мг ентекавіру для 147 із 149 пацієнтів у 3-му році, для всіх пацієнтів у 4 та 5-му році, та комбіновану терапію ентекавіром-ламівудином (з подальшою тривалою терапією ентекавіром) для середнього значення 20 тижнів для 130 із 149 пацієнтів у 3 -му році та протягом 1 тижня для 1 з 121 пацієнта у 4 -му році у дослідженні з перекиданням.
b Включає пацієнтів з принаймні одним вимірюванням ДНК HBV на етапі лікування методом ПЛР через 24 тижні або до 58 тижнів (рік 1), після 58 тижнів до 102 тижнів (рік 2), через 102 тижні до 156 тижнів (рік 3 ), після 156 тижнів до 204 тижнів (рік 4) або після 204 тижнів до 252 тижнів (рік 5).
c Пацієнти, яким також замінили LVDr.
збільшення ≥ 1 log10 вище надиру в ДНК HBV за допомогою ПЛР, що підтверджується наступними вимірами або наприкінці вікна часу.
Заміни ETVr (на додаток до заміни LVDr rtM204V / I ± rtL180M) спостерігалися на початковому етапі у ізолятах від 10/187 (5%) пацієнтів, стійких до ламівудину, які отримували ентекавір та контролювалися на резистентність, що свідчить про те, що попереднє лікування ламівудином може вибрати ці заміни резистентності і що вони можуть існувати з низькою частотою до початку лікування ентекавіром. Протягом 240 тижнів у 3 з 10 пацієнтів спостерігався вірусологічний рикошет (збільшення ≥ 1 log10 вище надиру). Початок резистентності до ентекавіру у дослідженнях пацієнтів, стійких до ламівудину протягом 240 тижнів, узагальнено у таблиці нижче.
Результати відображають використання комбінованої терапії ентекавіром-ламівудином (з подальшою тривалою терапією ентекавіром) у середньому 13 тижнів для 48 з 80 пацієнтів у 3-річному дослідженні, середня 38 тижнів для 10 із 52 пацієнтів у 4- рік та 16-тижневе дослідження для 1 з 33 пацієнтів у 5-річному дослідженні з перекиданням.
b Включає пацієнтів з принаймні одним вимірюванням ДНК HBV на етапі лікування за допомогою ПЛР на 24 тижні або після цього до 58 тижнів (рік 1), після 58 тижнів до 102 тижнів (рік 2), після 102 тижнів до 156 тижнів (рік 3), після 156 тижнів до 204 тижнів (рік 4) або після 204 тижнів до 256 тижнів (рік 5).
c Пацієнти, яким також проводили заміну LVDr.
збільшення ≥ 1 log10 вище надиру в ДНК HBV за допомогою ПЛР, що підтверджується наступними вимірами або наприкінці вікна часу.
eПочаток опору ETV щороку; вірусологічний відскок / рік.
Серед пацієнтів, резистентних до ламівудину, з ДНК HBV
05.2 "Фармакокінетичні властивості -
Поглинання: ентекавір швидко всмоктується з максимальною концентрацією у плазмі крові від 0,5 до 1,5 години. Абсолютна біодоступність не визначена. На основі екскреції незміненого препарату з сечею біодоступність оцінюється щонайменше у 70%. Після багаторазового дозування 0,1-1 мг спостерігається пропорційне збільшення значень Cmax та AUC. Рівноважний стан досягається через 6-10 днів після дозування один раз на день з ≈ 2 -кратним часом накопичення. Стаціонарні Cmax та Cmin становлять 4,2 та 0,3 нг / мл для дози 0,5 мг та 8,2 та 0,5 нг / мл для дози 1 мг відповідно. Таблетки та пероральний розчин були еквівалентними у здорових осіб; тому обидві лікарські форми можна міняти місцями.
Введення 0,5 мг ентекавіру зі стандартною їжею з високим вмістом жиру (945 ккал, 54,6 г жиру) або легкою їжею (379 ккал, 8,2 г жиру) призвело до мінімальної затримки всмоктування (1 - 1,5 години на повний шлунок проти 0,75 години натщесерце), зниження Cmax на 44 - 46% та зниження AUC на 18 - 20%. Зниження Cmax та AUC з їжею не вважається клінічно значущим у пацієнтів, які не отримували нуклеозидів, але може вплинути на ефективність у пацієнтів, що не мають резистентності до ламівудину (див. Розділ 4.2).
Розповсюдження: Орієнтовний об’єм розподілу ентекавіру перевищує загальну кількість води в організмі. Зв’язування плазми з білками сироватки людини в пробірці è ≈ 13%.
Біотрансформація: ентекавір не є субстратом, інгібітором або індуктором ферментної системи CPYP450. Після введення 14С-ентекавіру не спостерігалося окислювальних або ацетильованих метаболітів та незначної кількості метаболітів фази II, глюкуроніду та його сульфатних кон’югатів.
Ліквідація: ентекавір виводиться переважно нирками із сечею у незміненому стані у рівноважному стані приблизно 75% дози. Нирковий кліренс не залежить від дози і коливається від 360 до 471 мл / хв, що свідчить про те, що ентекавір піддається як клубочковій фільтрації, так і різній канальцевій секреції. Після досягнення пікових рівнів концентрація ентекавіру в плазмі знизилася бі-експоненціально з кінцевим періодом напіввиведення ≥ 128-149 годин. Спостерігається індекс накопичення препарату ≈ 2 рази при одноразовому добовому дозуванні, що свідчить про ефективний період напіввиведення приблизно 24 години.
Захворювання печінки: Фармакокінетичні показники у пацієнтів з помірним або важким захворюванням печінки подібні до таких у пацієнтів з нормальною функцією печінки.
Ниркова недостатність: Кліренс ентекавіру зменшується зі зменшенням кліренсу креатиніну.За 4-годинний сеанс гемодіалізу ≈ 13% дози було видалено та 0,3% видалено за допомогою CAPD.Фармакокінетичні дані ентекавіру після одноразової дози 1 мг (у пацієнтів без хронічного гепатиту В інфекції) наведені в таблиці нижче:
Трансплантація печінки: Експозиція ентекавіру у пацієнтів, інфікованих ВГВ, які перенесли трансплантацію печінки, які отримували стабільну дозу циклоспорину А або такролімусу (n = 9), була у ≥ 2 разів більшою за експозицію у здорових осіб з нормальною функцією нирок. Порушення функції нирок сприяло збільшенню експозиції ентекавіру у цих пацієнтів (див. розділ 4.4).
Секс: AUC була на 14% вищою у жінок, ніж у чоловіків, через відмінності у функції нирок та вазі. Після коригування відмінностей у кліренсі креатиніну та масі тіла не було відмінностей у експозиції між чоловіками та жінками.
Літні громадяни: Вплив на фармакокінетику ентекавіру на вік оцінювали шляхом порівняння людей похилого віку з віком від 65 до 83 років (середній вік у жінок 69 років, у чоловіків 74) з молодими людьми у віці від 20 до 40 років (середній вік у жінок 29 років, у чоловіків 25). AUC був на 29% вищим у літніх людей, ніж у молодих, головним чином через відмінності в функції нирок та вазі. Після коригування різниці у кліренсі креатиніну та масі тіла літні суб’єкти показали вищу AUC. Високу на 12,5% молодих суб’єктів . Фармакокінетичний аналіз населення, включаючи пацієнтів віком від 16 до 75 років, не показав, що вік істотно впливає на фармакокінетику ентекавіру.
Гонка: Фармакокінетичний аналіз населення не продемонстрував, що раса суттєво впливає на фармакокінетику ентекавіру.Однак висновки можна зробити лише для кавказької та азіатської груп, оскільки було небагато суб’єктів з інших категорій.
Педіатричне населення: Фармакокінетичні стадії рівноважного стану ентекавіру оцінювались (дослідження 028) у HBeAg-позитивних педіатричних пацієнтів віком від 2 до 18 років із компенсованою хворобою печінки, з яких 24 ніколи не лікувалися нуклеозидами, а 19 раніше лікувалися ламівудином. Експозиція ентекавіру серед суб’єктів, які не отримували нуклеозидів, що отримували добову дозу ентекавіру від 0,015 мг / кг до максимум 0,5 мг, була подібною до експозиції, досягнутої у дорослих, які отримували добову дозу 0,5 мг. Cmax, AUC (0-24) та Cmin у цих суб'єктів становили відповідно 6,31 нг / мл, 18,33 нг год / мл та 0,28 нг / мл.
Експозиція ентекавіру у пацієнтів, які раніше отримували ламівудин та отримували ентекавір у добовій дозі 0,030 мг / кг до максимальної дози 1,0 мг, була подібною до експозиції, досягнутої у дорослих, які отримували дозу 1,0 мг на добу. Cmax, AUC (0-24) та Cmin у цих суб'єктів становили відповідно 14,48 нг / мл, 38,58 нг год / мл та 0,47 нг / мл.
05.3 Доклінічні дані про безпеку -
У токсикологічних дослідженнях із повторними дозами на собаках спостерігалося «оборотне периваскулярне запалення центральної нервової системи. Однак це запалення не було виявлено у дозах, що в 9 і 10 разів перевищували дозування для людей (при введенні доз 0,5 і 1 мг) Цей ефект не спостерігалося у дослідженнях повторних доз на інших видах, включаючи мавп, які отримували ентекавір щодня протягом 1 року у дозах, що ≥ 100 разів перевищували дози, введені людям.
У дослідженнях з репродуктивної токсикології, в яких тваринам давали ентекавір протягом 4 тижнів, втрата фертильності у високих дозах самців або самок не спостерігалася. Зміни в яєчках (дегенерація насіннєвих трубчастих труб) спостерігалися при токсикологічних дослідженнях з повторною дозою на гризунах та собаках у дозах, що в 26 разів перевищували дози, що вводяться людям. Зміни в яєчках не спостерігалися в однорічному дослідженні мавп. У вагітних щурів та кроликів, яким вводили ентекавір, фактичний рівень ембріотоксичності чи токсичності для матері не відповідав дозам, що ≥ 21 разів перевищували дози, введені людям. У високих дозах у щурів спостерігалися такі ефекти: токсичність для матері, токсичність для ембріона та плоду (резорбція), зменшення маси тіла плода,
хвоста і хребців, зменшення окостеніння (хребців, грудин і фаланг) та додаткових поперекових хребців. У високих дозах у кроликів спостерігалися такі ефекти: ембріонально-фетальна токсичність (резорбція), зменшення окостеніння (під’язикової кістки), збільшення випадків 13-го ребра. Побічних явищ у нащадків не спостерігалося. В окремому дослідженні, в якому ентекавір вводили у дозі 10 мг / кг вагітним та годуючим самкам щурам, спостерігалося як вплив ентекавіру на плод, так і секреція ентекавіру в молоці. У молодих щурів, які отримували ентекавір з четвертого по вісімдесятий день після пологів, помітно знижена реакція на акустичні подразники спостерігалася протягом періоду відновлення (110-114 днів після пологів), але не протягом періоду введення при значеннях AUC ≥ 92 разів вище ніж у людей у дозі 0,5 мг або еквівалентній педіатричній дозі. Враховуючи границю впливу, цей висновок вважається малоймовірним з клінічної точки зору.
Генотоксичність не спостерігалася ні при тесті на мікробну мутагенність Еймса, ні при тесті генної мутації клітин ссавців, ні при трансформації ембріональних клітин сирійського хом'яка. І мікроядерне дослідження, і дослідження репарації ДНК у щурів були негативними. Ентекавір був кластогенним у культурах лімфоцитів людини у концентраціях, які були значно вищими, ніж ті, що були досягнуті в клінічних умовах.
У дворічних дослідженнях канцерогенності: у самців мишей збільшення випадків раку легенів у дозах ≥ 4 та ≥ 2 рази у дозах у людей у дозах 0,5 мг та 1 мг відповідно. Розвитку пухлини передувала проліферація пневмоцитів у легенях, що, однак, не спостерігалося у щурів, собак чи мавп, що свідчить про те, що ключовою подією у розвитку раку легенів у мишей, ймовірно, є видоспецифічний. спостерігається при тривалому введенні: збільшення випадків інших видів пухлин, включаючи гліому мозку у самців та самок щурів, рак печінки у самців мишей, доброякісні судинні пухлини у самок мишей та аденоми та карциноми печінки у самок щурів. Однак точний вплив на рівні не може бути встановлений. Передбачуваність таких спостережень у людей невідома.
06.0 ФАРМАЦЕВТИЧНА ІНФОРМАЦІЯ -
06.1 допоміжні речовини -
Ядро планшета:
кросповідон
моногідрат лактози
стеарат магнію
мікрокристалічна целюлоза
повідон
Покриття планшета:
диоксид титану
гіпромелоза
макрогол 400
Полісорбат 80 (E433)
06.2 Несумісність "-
Не актуально.
06.3 Строк дії "-
2 роки
06.4 Особливі умови зберігання -
Пухир:
Не зберігати при температурі вище 30 ° C. Зберігати в оригінальній коробці.
Пляшки:
Не зберігати при температурі вище 25 ° C. Тримайте пляшку щільно закритою.
06.5 Характер безпосередньої упаковки та вміст упаковки -
Кожна коробка містить:
§ 30 х 1 таблетка, вкрита плівковою оболонкою; 3 блістери по 10 х 1 таблетки, вкритій плівковою оболонкою, кожен перфорований одиничний дозу алюмінію / алюмінію або:
§ 90 х 1 таблетка, вкрита плівковою оболонкою; 9 блістерів по 10 х 1 таблетці, вкритій плівковою оболонкою, у кожному блістері з перфорованою алюмінію / алюмінію
Пляшка з поліетилену високої щільності (HDPE) з поліпропіленовою дитячою кришкою, що містить 30 таблеток, вкритих оболонкою. Кожна коробка містить одну пляшку.
Не всі розміри упаковок та типи контейнерів можуть продаватися.
06.6 Інструкції з використання та поводження -
Невикористані ліки та відходи, отримані з цього препарату, слід утилізувати відповідно до місцевих правил.
07.0 ВЛАСНИК "РОЗРОБНИЦТВА"
BRISTOL-MYERS SQUIBB PHARMA EEIG
Бізнес -парк Аксбридж
Сандерсон -роуд
Uxbridge UB8 1DH
Великобританія
08.0 НОМЕР РОЗВИТКУ З РОБОТИ
Пухир: EU/1/06/343/003
037221076
ЄС/1/06/343/006
Пляшка: EU/1/06/343/001
037221052
09.0 ДАТА ПЕРШОГО ДОЗВІЛЕННЯ АБО ОНОВЛЕННЯ ДОЗВІЛА -
Дата першого дозволу: 26 червня 2006 року
Дата останнього оновлення: 26 червня 2011 р
10.0 ДАТА ПЕРЕГЛЯДУ ТЕКСТУ -
D.CCE, серпень 2014 р