Спостерігається у одного з кожних 100 000 новонароджених, синдром Пфайффера асоціюється з мутацією генів FGFR1 та FGFR2; обидва ці гени мають завдання регулювати зрощення черепних швів та розвиток пальців рук і ніг.
Для діагностики синдрому Пфайффера фундаментальним є фізикальне обстеження, анамнез, рентгенологічна оцінка черепа, пальців рук і ніг і, нарешті, генетичний тест.
В даний час ті, хто страждає на синдром Пфайффера, можуть розраховувати лише на симптоматичне лікування, тобто на те, що полегшує симптоми.
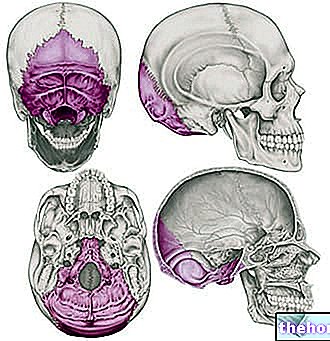
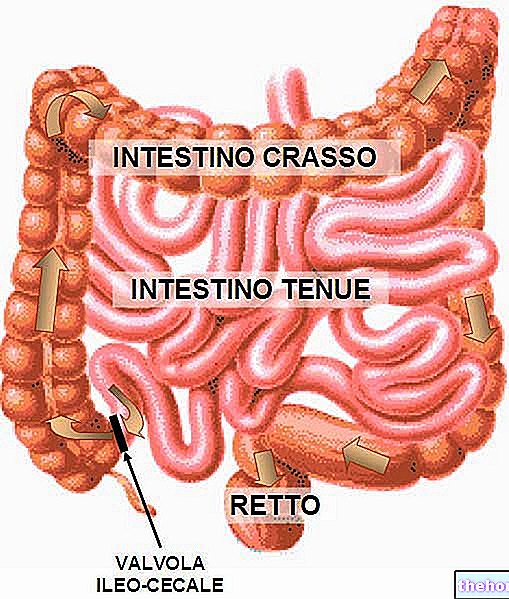
Короткий огляд черепних швів та їх зрощення
Черепні шви - це фіброзні суглоби, які служать для злиття кісток черепного склепіння (тобто лобової, скроневої, тім'яної та потиличної кісток).
У нормальних умовах процес зрощення черепних швів проходить у постнатальний період, починаючи з 1-2-річного віку для деяких суглобових елементів і закінчуючи у 20-річному віці для інших. Цей тривалий і частотний процес злиття дозволяє мозку рости і адекватно розвиватися.
- Наявність аномально великих і відхилених великих і великих пальців ніг таким чином, що вони, здається, віддаляються від інших пальців (медіальне відхилення).
Отже, синдром Пфайффера - це генетичний стан, який у тих, хто його носить, в основному визначає аномалії в черепі та руках.
Оскільки читачі матимуть можливість дізнатися більше у розділі, присвяченому симптомам, однак, синдром Пфайффера може бути пов’язаний з іншими проблемами та іншими фізичними вадами розвитку.
Епідеміологія: наскільки поширений синдром Пфайффера?
За статистикою, кожен сильний синдром Пфайффера народжується на кожні 100 000 осіб.
Ви знали, що ...
Генетичних захворювань, які, як і синдром Пфайффера, викликають краніосиностоз, близько 150.
Серед них, на додаток до синдрому Пфайффера, важливістю виділяються синдром Крузона, синдром Аперта та синдром Зетра-Чотцена.
Що викликає мутацію гена, пов'язану з синдромом Пфайффера?
Приміщення: гени, присутні в хромосомах людини, є послідовностями ДНК, які мають завдання виробляти основні білки в біологічних процесах, необхідних для життя, включаючи ріст і реплікацію клітин.
Коли вони вільні від мутацій (отже, у здорової людини), гени FGFR1 та FGFR2 виробляють у належних кількостях, відповідно, рецептор фактора росту фібробластів 1 та рецептор фактора росту фібробластів 2, які є двома рецепторними білками, необхідними для позначення терміни зрощення черепних швів і регулювання розвитку пальців рук і ніг (іншими словами, вони сигналізують, коли настає відповідний час для зрощення черепних швів і контролюють формування пальців і стоп).
З іншого боку, коли вони зазнають мутацій, що спостерігаються за наявності синдрому Пфайффера, гени FGFR1 і FGFR2 є гіперактивними і виробляють вищезгадані рецепторні білки в такій величезній кількості, що час злиття черепних швів змінюється (вони швидше ) і процес тренування пальців рук і ніг відбувається неправильно.
Синдром Пфайфера - аутосомно -домінантне захворювання
Зрозуміти...
Кожен ген людини присутній у двох копіях, які називаються алелями, одна з материнського походження та одна з батьківського походження.
Синдром Пфайффера має всі ознаки аутосомно -домінантного захворювання.
Генетичне захворювання є аутосомно -домінантним, коли мутації однієї копії гена, що викликає його, достатньо, щоб проявити себе.
Види синдрому Пфайффера
У 1993 році, після численних досліджень синдрому Пфайффера, американський лікар Майкл Коен опублікував типологічну класифікацію генетичної хвороби, про яку йдеться, яка передбачала існування трьох патологічних варіантів, ідентифікованих просто термінами "тип I", "тип II" і Тип III "і всі поділяють наявність краніосиностозу та аномалій великого пальця і великих пальців ніг. Медико-наукове співтовариство негайно прийняло цю класифікацію, і з тих пір фахівці з синдромом Пфайффера використовували її як діагностичний інструмент та для оцінки тяжкості наявного генетичного стану; насправді, слід зазначити, що класифікація доктора Коена розрізняє синдром Пфайффера на основі тяжкості черепних та цифрових аномалій та наявності інших симптомів та ознак.
Переходячи до деталей окремих патологічних варіантів, на цьому етапі статті важливо підкреслити, що:
- The Тип I. це менш важка версія синдрому Пфайффера, оскільки краніостеноз та аномалії великого і великого пальців ніг мають обмежені наслідки.
Інша важлива інформація: це обумовлено мутацією FGFR2, іноді поєднаною з мутацією FGFR1; це може бути спадковий або набутий стан. - The Тип II це найважчий варіант синдрому Пфайффера, оскільки він асоціюється з важким краніосиностозом, майже несумісним з життям, та з глибокими відхиленнями в руках і ногах.
Інша важлива інформація: це виключно через мутацію FGFR2; це завжди набутий стан. - The Тип III це версія синдрому Пфайффера, яка за шкалою тяжкості падає трохи нижче типу II, але значно вище типу I, оскільки нинішній краніосиностоз майже настільки ж важкий, як і варіант, описаний у попередньому пункті.
Інша важлива інформація: це обумовлено виключно мутацією FGFR2; це завжди набутий стан.
Краніостеноз
У носіїв синдрому Пфайфера краніосиностоз може, залежно від кількості черепних швів, залучених у процес раннього зрощення, мати такі наслідки:
- Абсолютно аномальний вертикальний розвиток голови у поєднанні з відсутністю бічного розширення черепа. Отже, у пацієнта з синдромом Пфайффера довга вузька голова;
- Формування високого і видатного чола;

- Підвищення внутрішньочерепного тиску, від якого залежать такі симптоми, як постійний головний біль, проблеми із зором, блювота, дратівливість, проблеми зі слухом, проблеми з диханням, змінами психічного стану, набряком папіломи;
- Інтелектуальний дефіцит, що призводить до зниження IQ. Інтелектуальні недоліки є результатом зменшення простору для росту, яким користується мозок після передчасного зрощення корональних черепних швів;
- Відсутність розвитку проміжної частини обличчя, яка виглядає плоскою, якщо не увігнутою;
- Наявність опуклих (проптоз), широко відкритих і аномально розставлених очей (очний гіпертелоризм);
- Наявність дзьобастого носа;
- Порушення розвитку щелепи (гіпоплазія верхньої щелепи), що призводить до стану скупчення зубів;
- Конюшиноподібний вигляд голови ("череп конюшини"). "Череп конюшини" викликає гідроцефалію.
ТИП I
Синдром Пфайффера I типу асоціюється з легким клінічним краніосиностозом, який дуже часто обмежується наданням подовженої форми черепа та спричиненням помітно високого чола та плоского обличчя.
При правильному лікуванні люди з синдромом Пфайффера I типу зазвичай ведуть нормальний спосіб життя і мають нормальний коефіцієнт інтелекту.
ТИП II
Синдром Пфайффера II типу є єдиним патологічним варіантом, що викликає так званий "череп трійки", ця черепна аномалія має серйозні наслідки для інтелектуальних здібностей і часто асоціюється з передчасною смертю.
Ті, хто страждає синдромом Пфайффера II типу, представляють всю описану вище клінічну картину щодо наслідків краніосиностозу.

ТИП III
Синдром Пфайффера III типу має такий же вплив на його носіїв, як і синдром Пфайффера II типу, за винятком "черепа трилисника".
Люди з синдромом Пфайффера III типу не мають тривалої тривалості життя.
Аномалії, що вражають великі пальці рук і ніг
Якщо особливо серйозні, аномалії, що впливають на великі пальці рук і на великі пальці ніг, можуть глибоко погіршити функціональні можливості рук і ніг, викликаючи проблеми в захопленні предметів та / або ходьбі.
Ви знали, що ...
Медіальне відхилення, що впливає на великі пальці рук і на великі пальці ніг у пацієнтів з синдромом Пфайффера, є прикладом варус -варус. Точніше, лікарі говорять про варус великого пальця, обумовлений медіальним відхиленням великих пальців, і валлі галюкс, обумовлений медіальним відхиленням великих пальців ніг.

Брахідактилія
При синдромі Пфайффера брахідактилія є досить поширеною аномалією, яка може вражати лише кілька пальців або весь цифровий комплекс кистей та / або ніг.
Проблема брахідактилії спостерігається у всіх типологічних варіантах, хоча і з різною частотою.
Синдактилія
При синдромі Пфайффера синдактилія становить досить часту «аномалію (рідше, ніж брахідактилія), яка може мати різні конотації (вона може бути неповною, повною, складною тощо).
Проблема брахідактилії спостерігається у всіх типологічних версіях синдрому Пфайффера, хоча і з різними рецидивами.
Кістковий анкілоз
Синдром Пфайффера асоціюється, перш за все, з кістковим анкілозом ліктя, хоча насправді він може спричинити таку ж проблему для будь -якого великого суглоба в тілі людини.
Кістковий анкілоз - це проблема, яка зустрічається лише в найважчих типологічних варіантах синдрому Пфайффера (особливо при II типі).
Аномалії, що вражають дихальні шляхи
Можливі аномалії в дихальних шляхах, спричинені синдромом Пфайффера, такі, що спричиняють проблеми з диханням з серйозними наслідками для загального стану здоров’я пацієнта (найбільше страждає мозок).
Як і кістковий анкілоз, зазначені вище аномалії спостерігаються лише у більш важких типологічних варіантах (зокрема, типу II).
Коли можна виявити синдром Пфайффера?
Як правило, черепні та цифрові аномалії, спричинені синдромом Пфайффера, виявляються при народженні, тому діагностика та планування лікування є негайними.
до голови (рентген голови, КТ голови та / або МРТ голови) та до рук та ніг; нарешті, він закінчується генетичним тестом.
Фізичний огляд та історія хвороби
Фізичний огляд та анамнез по суті полягають у точній оцінці симптомів, які проявляє пацієнт.
У контексті синдрому Пфайффера, саме на цих фазах діагностичного процесу лікар встановлює краніостеноз та аномалії, що зачіпають великі пальці рук і великих пальців ніг, і, виходячи з інших наявних симптомів, висуває гіпотезу про типологічний варіант.
Рентгенологічне дослідження голови і пальців рук і ніг
У контексті синдрому Пфайффера,
- Рентгенологічні дослідження голови використовуються лікарем для підтвердження наявності раннього зрощення черепних швів та оцінки тяжкості черепно-мозкових аномалій.
- З іншого боку, рентгенологічні дослідження необхідні для дослідження ступеня варусу та "можливої брахідактилії та / або" можливої синдактилії.
Генетичний тест
Це аналіз ДНК, спрямований на виявлення мутацій у критичних генах.
У контексті синдрому Пфайффера він є підтверджуючим діагностичним тестом, оскільки дозволяє виділити мутацію FGFR2 та / або FGFR1.
Генетичний тест також є тестом, який дозволяє встановити тип наявного синдрому Пфайффера.


-cos-cause-e-terapia.jpg)